Case Report

Retroperitoneal Paraganglioma: Clinical Presentation and Treatment Outcomes in Two Patients.
1García Marchiñena Patricio, 1Jurado Alberto, 1Uria Luis, 1Costabel José, 2Mariana Isola, 1Damia Oscar, 1Gueglio Guillermo.
1 Urology Department, Hospital Italiano de Buenos Aires. 2 Pathology Department, Hospital Italiano de Buenos Aires.
Submitted Tuesday, July 16, 2013
Accepted Tuesday, September 10, 2013
Published Friday, October 18, 2013
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Introduction
Paragangliomas are rare neuroendocrine tumors that arise from extra-adrenal autonomic ganglia. The aim of this paper is to analyze the presentation, diagnosis and treatment outcomes of two patients with retroperitoneal paraganglioma treated in our hospital.
Case Presentation
The first case is a 34 year old female, ASA status 1, who underwent elective surgery for removal of retroperitoneal tumor. Laparoscopic surgical resection was decided. Minimal tumor manipulation was followed with severe hypertension. Due to high suspicion of the presence of a catecholamine-secreting tumor, intravenous antihypertensive drugs were given. The patient coursed with hypotension and acute pulmonary edema after the surgery. Transthoracic echocardiogram showed apical ballooning with impaired ejection fraction of the left ventricle. After 3 days in Cardiac Care Unit the patient was discharged. The second case is a 25 years old male, with large history of refractory hypertension who was admitted in our hospital with sweating, palpitations, dizziness and hypertension. Catecholamine dosage was performed in plasma and urine and the results were inconclusive. The CT scan showed a right hypoplastic kidney and a 2.5 x 1.9 cm nodular mass located between his left renal artery and vein. Because of the high suspicion of paraganglioma we decided the tumor excision. The patient recovered without complications, and was discharged 4 days after surgery. Final surgical pathology demonstrated in both cases a paraganglioma.
Conclusions
The paraganglioma is a rare tumor associated with high morbidity and mortality when the diagnosis is made intraoperatively.
Key words
Extra-adrenal pheochromocytoma, Retroperitoneal mass, diagnosis.
Introduction
Paragangliomas are rare neuroendocrine tumors that arise from extra-adrenal autonomic ganglia (sympathetic or parasympathetic). They are closely related to pheochromocytomas (also called intra-adrenal paraganglioma) and are histologically indistinguibles [1]. Most of these tumors are hormonally active (up to 60 percent) and have a high incidence of malignancy (13 to 26 percent) [2]. The aim of this paper is to analyze the presentation, diagnosis and treatment outcomes of two patients with retroperitoneal paraganglioma located in the renal hilium treated in our hospital.
Case report
The first case is a 34 year old female, American Society of Anesthesiologist (ASA) status 1, underwent elective surgery for removal of retroperitoneal tumor. Our patient have no important clinical history, ande the tumor was discovered during a routine examination. Before the surgery, she underwent a percutaneous biopsy of the lesion, which courses without complications, reporting the pathological study insufficient material. The CT scan showed a 6x4x 3.5 cm latero aortic solid mass which contacts with left renal hilum. She underwent a percutaneous biopsy of the lesion, which courses without complications, reporting the pathological study insufficient material. With a diagnosis of retroperitoneal mass of unknown origin, the laparoscopic surgical resection was decided. Preoperative findings, electrocardiogram and blood pressure measured were normal. Retroperitoneoscopic access was performed using previously described technique3. Minimal tumor manipulation was followed with severe hypertension measuring 320/110 mmHg and tachycardia up to 120 beats/minute. Surgery was temporally interrupted, until hemodynamically compensation of the patient. Due to high suspicion of the presence of a catecholamine-secreting tumor, intravenous dose of propranolol and phentolamine was given. Once the tumor was excised the patient coursed with hipotension and acute pulmonary edema. Transthoracic echocardiogram showed hypokinesia of medial and apical segments (apical ballooning) with moderately impaired ejection fraction of the left ventricle. A coronary angiography was performed post operatively to rule out coronary lesions. The patient improved with decreased inotropic requirements and was discharged at third day of the surgery. The pathological diagnosis (see Figure 1)was retroperitoneal paraganglioma with negative surgical margins. We performed a positron emission tomography (PET / CT FDG) whole body, for staging, with no evidence in this study associated injuries. One year after surgery the patient is free of disease and symptoms.
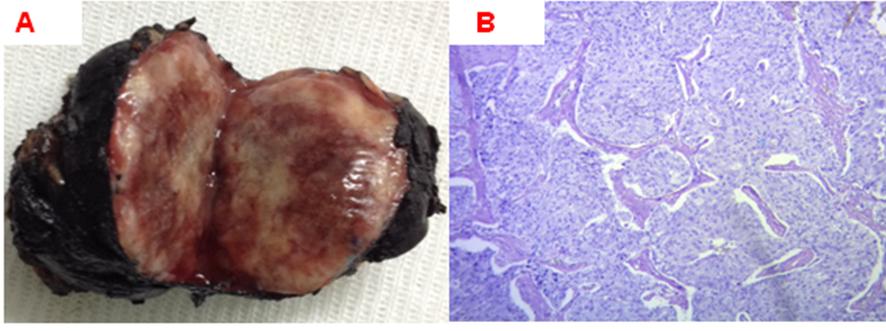
Figure 1. Pathology: paraganglioma. Macroscopy (A) tumor nodule brownish lobulated surface and soft consistency of 60 x 45 x 31 mm. Microscopy (B) tumor proliferation of spindle cells with ovoid nuclei chromatin stippling "salt and pepper" and large finely granular basophilic cytoplasm.
The second case is a 25 years old male, with large history of hypertension refractory to medical treatment who was admitted in our hospital with sweating, palpitations, dizziness and high blood pressure records 180/120mmHg. Antihypertensive treatment was begun with intravenous nitroprusside. Once the patient was hemodynamically stable, oral antihypertensive treatment with four drugs started (doxazosin, propranolol, nebivolol and nifedipine). Catecholamine dosage was performed in plasma and urine and the results were inconclusive. The CT scan showed a right hypoplastic kidney and a 2.5 x 1.9 cm nodular mass located between his left renal artery and vein (see Figure 2).
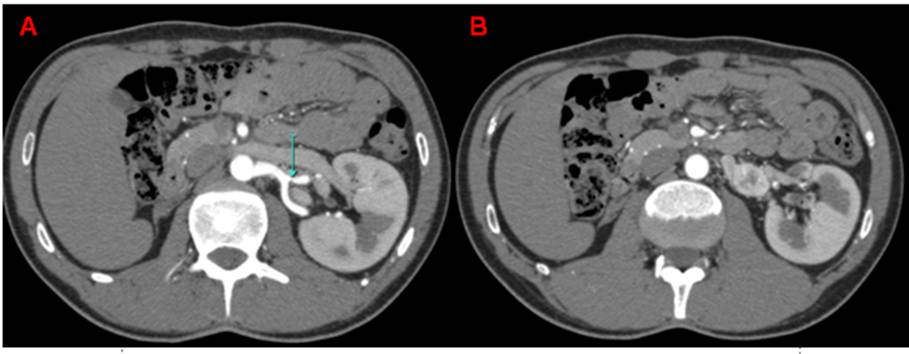
Figure 2. CT scan shows an hypertrophied left kidney. (A) Just below the renal artery a nodular formation. (B) caudal extension laterally to the aorta (B).
The metaiodobenzylguanidine scintigraphy was normal without uptake at the level of the lesion. We decided to make the tumor excision by open surgery through a left subcostal incision. The patient recovered without complications during both intra-and postoperatively. Discharge was given 4 days after surgery with 3 antihypertensive drugs. Final surgical pathology demonstrated 2,2x1,5x0,4 cm paraganglioma.
Discussion
Pheochromocytomas are rare tumors usually arising from the adrenal medulla. In 14% to 25% of cases, they may arise from embryological adrenal remnants referred as extra-adrenal paragangliomas [4]. The estimated annual incidence of pheochromocytoma / paraganglioma is 0.8 per 100,000 people per year. Most paragangliomas are diagnosed in the third to fifth decades of life. Patients with hereditary paragangliomas tend to develop disease a decade earlier than do those with sporadic disease. The male to female ratio is equal in patients with hereditary paraganglioma, while sporadic tumors are much more common in women (71% versus 29%) [5]. In addition to variability in relation to location, paragangliomas refute the "10% rule" applied to pheochromocytomas, since about 26% of paragangliomas are multiple, 30-50% are associated with a hereditary syndrome and about 20% of paragangliomas secreting (abdominal and mediastinal) are malignant [6]. The four types of presentation that describes, in descending order of frequency are: mass effect (usually the head and neck), hypersecretion of catecholamines, asymptomatic and an incidental finding on imaging study [7]. Biochemical diagnosis of paraganglioma is done by measurements fractionated metanephrines and catecholamines in urine and / or plasma. Imaging studies provide information about the location of the tumor, vascularization and relationship to adjacent organs. The methods used are computed tomography (CT) and magnetic resonance imaging (MRI). If the CT or abdominal and pelvic MRI are negative in the presence of clinical and biochemical evidence of a catecholamine-secreting tumor, the next step is to apply functional studies with radioisotopes images using metaiodobenzylguanidine (MIBG) or 18F-fluoro-2-deoxyglucose (FDG) -PET [8]. The differential diagnosis of an abdominal mass includes carcinomas, lymphomas and soft tissue sarcomas, including gastrointestinal stromal tumors and desmoid tumors. The definitive diagnosis is achieved at the time of resection. A biopsy (incisional or fine needle aspiration) is contraindicated in a patient with suspected paraganglioma because it can trigger a severe hypertensive crisis punctures can be confused with many different malignancies [9-10].
Once a paraganglioma is suspected, the patient should undergo surgical resection after adequate medical preparation. Surgical resection is a high-risk procedure that requires an experienced team (surgeon / anesthesiologist). Preoperative medical treatment is to control blood pressure (including the prevention of hypertensive crisis during surgery). The medical preparation with alpha and beta blockers is indicated in all preoperative patients with catecholamine-secreting tumors in order to control intraoperative blood pressure and prevent hypertensive crises. Even with modern genetic testing, most paragangliomas appear to be sporadic [11]. Paragangliomas most have been linked to inherited mutations in the genes that encode different subunits of succinate dehydrogenase (SDH). Furthermore, pheochromocytoma /paraganglioma are a component of four genetic syndromes: multiple endocrine neoplasia type 2A and 2B (MEN2), neurofibromatosis type 1 (NF1), von Hippel Lindau disease (VHL), and Carney-Stratakis dyad. Most cases of hereditary paraganglioma posted by mutations in SDHD, SDHB and SDHC, VHL and NF1 [12].Genetic screening is recommended for all patients with a diagnosis of paraganglioma. The presence of multiple paragangliomas, diagnosis in childhood, history of associated tumors, family history and the location of the head and neck should make us consider hereditary disease [13].The histologic features such as nuclear pleomorphism, necrosis, mitotic index, local invasion and can be seen in benign paragangliomas are not diagnostic of malignancy. According to the World Health Organization 2004 (WHO) the only real indicator of malignant behavior metastatic spread is [14].
Conclusion
The paraganglioma is a rare tumor associated with high morbidity and mortality when the diagnosis is made intraoperatively. Is essential to rule out the existence of paraganglioma in all retroperitoneal tumors, even in asymptomatic and hemodynamically stable patients. Preoperative diagnosis helps to plan treatment and prevent complications.
Competing interests
The authors declare that there are no competing interests
Authors' Contribution
GMP: Concept and design, preparation of manuscript
JA: Concept and design, preparation of manuscript
UL: Concept and design, preparation of manuscript
CJ: Concept and design, preparation of manuscript
MI: Concept and design, preparation of manuscript
DO: Concept and design, preparation of manuscript
GG: Concept and design, preparation of manuscript
Acknowledgement
None
References
[1]1- Lin D, Carty SE, Young WF. Paragangliomas: Epidemiology, clinical presentation, diagnosis, and histology. [Monografía en Internet]. UpTodate; 2013 [acceso 2 de Mayo 2013]. Disponible en: http://www.uptodate.com/
[2]Tomulic K, Pavicic-Saric J, Kocman B, et al. Successful Management of unsuspected retroperitoneal paraganglioma via the use of combined epidural and general anesthesia: a case report. J Med Case Rep 2013;7(1):58.[pubmed]
[3]Jurado A, García Marchiñena P, Billordo Peres N, et al. Acceso retroperitoneoscópico para el manejo conservador de los tumores renales. Rev. Arg. de Urol. 2012;77(3):176-182.
[4]Lehrfeld T, Natale R, Sharma S, et al. Robot-Assisted Excision of a Rtroperitoneal Mass Between the Left Renal Artery and Vein. JSLS. 2010;14(3):447-9.[pubmed]
[5]Eisenhofer G, Timmers HJ, Lenders JW y col. Age at diagnosis of pheochromocytoma differs according to catecholamine phenotype and tumor location. J Clin Endocrinol Metab 2011; 96:375.[pubmed]
[6]Parenti G, Zampetti B, Rapizzi E y col. Update and new perspectives on diagnosis, prognosis, and therapy of malignant pheochromocytoma/paraganglioma. J Oncol 2012; 2012: 872713.[pubmed]
[7]Erikson D, Kudva YC, Ebersold MJ, y col. Benign paragangliomas: clinical presentation and treatment outcomes in 236 patients. J Clin Endocrinol Metab 2001; 86:5210.[pubmed]
[8]Chen, Sippel RS, O´Dorisio MS, y col. The North American Neuroendocrine Tumor Society Consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroidcancer. Pancreas 2010; 39:775.[pubmed]
[9]Vanderveen KA, Thompson SM, Callstrom MR, y col. Biopsy of pheochromocytomas and paragangliomas: potential disaster. Surgery 2009; 146: 1158.[pubmed]
[10]Kubota K, Kato S, Mawatari H, y col. Risky endoscopic ultrasonography-guided fine-needle aspiration for asymptomatic retroperitoneal tumors. Dig Endosc 2010; 22:144.[pubmed]
[11]Tauzin-Fin P, Sesay M, Gosse P, y col. Effects of perioperative alpha 1 block on haemodynamic control during laparoscopic surgery for phaeochromocytoma. Br J Anesth 2004; 92:512.[pubmed]
[12]Welander J, Söderkvist P, Gimm O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer 2011; 18: 253.[pubmed]
[13]Benn DE, Gimenez-Roqueplo A, Reilly JR, y col. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab 2006; 91:827.[pubmed]
[14]Kimura N, Watanabe T, Noshiro T, y col. Histological gradind of adrenal and extra-adrenal pheochromocytomas and relationship to prognosis: a clinicopathological analysis of 116 adrenal pheochromocytomas and 30 extra-adrenal sympathetic paragangliomas including 38 malignant tumors. Endocr Pathol 2005; 16:23.

