Case Report

Extraskeletal Ewings Sarcoma Presenting as Nasopharyngeal Mass- Rare Tumour
and Rarest Presentation
*Mona Bargotya, *Kiran Agarwal, *Reema Bhushan, *Archna Rautela Pahwa
- Submitted: February 01, 2014
- Accepted:February 07, 2014
- Published:March 16, 2014
This is an Open Access article distributed under the terms of the Creative Commons Attribution License ((http://creativecommons.org/licenses/by/3.0)which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited
Abstract
Extraskeletal Ewing’s sarcoma (EES) is a rare, rapidly
growing, round-cell, malignant tumor of uncharacterized mesenchymal cell origin
that can develop in the soft tissues at any location and is morphologically
similar to the commoner Ewing’s sarcoma arising from bone. EES can develop in
soft tissues of any location but its occurrence in head and neck as a primary
tumor is very unusual. It occurs predominantly in adolescents and young adults
between 10-30 years of age and it follows an aggressive course with a high rate
of recurrence.
Introduction
Extaskeletal Ewing’s sarcoma (EES) first described by Tefft
et al., [1] is a very rare, rapidly growing malignant round
cell tumor of uncharacterized mesenchymal origin which histologically resembles
Ewing’s sarcoma and can develop in the soft tissues at any location, usually in
the lower extremities, paravertebral region, chest wall, retroperitonium
[2,3]. EES of the head and neck region
is very rare and accounts for only 2-3% of all the cases [4, 5]
and most of the cases being reported in the maxillary sinus followed by ethmoid
sinus. Young adults and age group between 10-30 years is usually affected but
cases have been reported between 14 months and 63 years of age [6, 7].
We herein present a case of EES presenting as nasopharyngeal mass in a 21 year old female.
Case Report
A 21-year-old female presented with rapidly growing mass
over soft palate and history of nasal obstruction for 2 years with difficulty in
swallowing since 1 year. On oral examination, a pinkish globular non-tender firm
mass involving the soft palate measuring 4x3 cm in size with small ulcerations
in hard palate were seen. Nasal examination revealed a mass visible in the right
nasal cavity extending to the right side of the nasopharynx. CT scan (Figure 1)
showed a soft tissue heterogeneous density lesion measuring 11x5x4 cm in the
right nasopharynx involving adjacent oropharynx with involvement of soft palate
and destruction of right medial pterygoid plate.
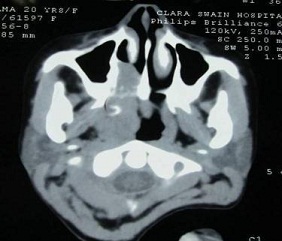
Fig 1: CECT- Scan- Heterogenous soft tissue density measuring 11x5x4 cms
Fine needle aspiration was done from the soft palate mass
and smears obtained were richly cellular showing loosely cohesive as well as
dispersed uniform, small round cells with scant cytoplasm, and indistinct cell
borders. Nuclei were round having fine nuclear chromatin and inconspicuous
nucleoli.
Occasional pseudorosette formation was also seen. On this
cytomorphological basis a cytological diagnosis of small round cell tumor was
being kept and biopsy was advised. Histopathological sections revealed focally
ulcerated overlying epithelium and with underlying stroma showing tumor cells in
sheets with focal peritheliomatous pattern. (Figure 2) The
tumor cells were round to oval in shape with scant to moderate amount of
eosinophilic cytoplasm and occasional cells showed vacuolization. The cells
showed mild anisonucleosis with round to oval nucleus having fine granular
chromatin with 0-1 inconspicuous nucleoli. Mitotic count was 0-1/HPF. In
addition, the tumor cells also showed strong positivity for PAS stain. On the
basis of histopathological examination differential diagnosis of Ewing’s
sarcoma, PNET, neuroblastoma or rhabdomyosarcoma were suggested and
immunohistochemical panel for vimentin, desmin, chromogranin and CD99 was put.
The present case showed strong positivity for CD99 (Figure 3) and vimentin but was however negative for desmin and chromogranin.
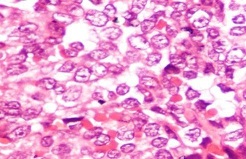
Fig 2: (H&Ex400) Tumour cells with scant to moderate amount of cytoplasm, round to oval nuclei with mild anisonucleosis, fine granular chromatin and 0-1 inconspicuous nucleoli
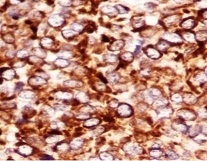
Fig 3: Strong CD 99 positivity in tumour cells
On the basis of histological, immunohistochemical, clinical
and radiological findings a final diagnosis of Extraskeletal Ewing’s sarcoma was
made. Following the diagnosis, the patient was immediately put on chemotherapy
as EES is a radiosensitive tumor. After 2 cycles of chemotherapy the tumor size
decreased significantly and the patient responded very well to therapy with
improvement in her general condition. Post chemotherapy period has been
uneventful till date..
Discussion
EES in head and neck region accounts for only 2-3% of all
Ewing’s sarcomas and is exceptionally rare in nasal region. It responds well to
chemotherapy, however has a grave prognosis with high rate of recurrence and
metastasis commonly to lung and bone. Although rare it should be considered in
differential diagnosis in young adults presenting with large heterogeneous mass
in head and neck region.
Wide spectrum of small round cell tumors in the sinonasal
region which include metastatic neuroblastoma, rhabdomyosarcoma and PNET can
pose significant diagnostic challenges hence careful clinical, radiological
evaluation and microscopic features along with Immunohistochemistry (IHC) and
cytogenetics help in reaching an accurate diagnosis and appropriate management.
Neuroblastoma is usually seen in younger age group and shows the presence of
Homer-Wright rosettes with elevated urinary catecholamine metabolite level and
presence of neuropil and ganglionic differentiation. The cells of neuroblastoma
are strongly positive for NSE and are negative for CD99.Rhabdomyosarcoma show
small round cells with small nucleoli and presence of eosinophilic cells
characteristic of rhabdomyoblast with or without cross striations. Most of the
rhabdomyosarcomas show positive immunostaining for myogenic markers including
myogenin and Myo- D1. Histological evidence of rosette formation along with
immunohistochemical evidence of neural differentiation is required for the
diagnosis of PNET. There have been recent advancements in immunohistochemistry
which have further aided in the diagnosis of EES. Since the expression of the
cell surfaces glycoprotein p30/32 on the EES tumor cells can be recognized by
monoclonal antibody O-13, small round tumor with strong immunoreactivity for
O-13 is highly indicative of EES [8, 9].
Although neuroblastoma, rhabdomyosarcoma and PNET may occasionally show positive
staining by O-13 antibody [9], their respective diagnosis
can be eliminated by negative staining for chromogranin, neurofilament, HHF-13,
desmin and myogenin.
Although the prognosis of EES is grave, it still remains a
potentially curable tumor. Chemotherapy is highly effective in reducing the
tumor size as well as clearing micrometastasis which is invariably present in
80% of the cases (10).
Conclusion
As EES has as such no specific clinical and radiological
features so proper and representative timely biopsy is the best method to
establish an accurate diagnosis as it is often confused with other small round
cell tumors. Hence, EES though a very rare entity should be considered when an
expansile, invasive nasopharyngeal mass is detected with destructive bony
changes.
Key Message
EES itself is very rare tumor and above all its presentation
in head and neck region is still rarer. Because of the rarity of EES, very few
clinical studies are available. Realizing the nature of Ewing’s sarcoma and
understanding its diagnostic significance can lead to the approach of
appropriate management.
Authors' Contribution
MB: Literature search and drafted
manuscript.
KA: Pathological diagnosis and edited final
manuscript.
RB: Interpreted result.
ARP:
Helped in drafting manuscript.
Conflict of Interests
The authors declare that there are no conflicts of interests.
Ethical Considerations
Written informed consent was obtained from the patient for
publishing this case report.
Funding
None Declared
Acknowledgement
None
References
[1].Tefft M, Vawter GF, Mitus A. Paravertebral “round cell tumours” in children. Radiology 1969; 92:1501-9.[pubmed]
[2].Gupta S. Gupta OP, Mehrotra S, Mehrotra D. Ewing’s sarcoma of the maxilla: A rare presentation. Quintessence Int 2009; 40.135-40. [pubmed]
[3].Coskun BU, Cinar U ,Savk H ,Basak T, Dadas B.Isolated maxillary sinus Ewing’s sarcoma. Rhinology 2005;43:225-228. [pubmed]
[4].Kawabata M, Yoshifuku K, Sagara Y, Kurono Y. Ewing’s sarcoma /primitive neuroectodermal tumour occurring in the maxillary sinus.Rhinology 2008;46:75-78. [pubmed]
[5].Afrezon M, Wood WE, Powell JR. Ewing’s sarcoma of the ethmoid sinus. Otolaryngol Head Neck Surg 2003; 128:897-901. [pubmed]
[6].Angervall L, Enzinger FM. Extraskeletal neoplasm resembling Ewing’s sarcoma .Cancer 1975; 36:240-51. [pubmed]
[7].Rud NP, Reiman HM, Pritchard DJ, Frassica FJ, Smithson WA. Extraosseous Ewing’s sarcoma: a study of 42 cases. Cancer 1989; 64:1548-53. [pubmed]
[8].Sexton CW, White WL. Primary cutaneous Ewing’s family sarcoma. Report of a case with immunostaining for glycoprotein p30/32 mic2. Am J Dermatopathol 1996; 18:601-5. [pubmed]
[9].Weidner N, Tjoe J. Immunohistochemical profile of monoclonal antibody O13: antibody that recognizes glycoprotein p30/32MIC2 and is useful in diagnosing Ewing’s sarcoma and peripheral neuroepithelioma. Am J Surg Pathol 1994; 18:486-94. [pubmed]
[10].Hafazi S, Seethala RR. Stelow EB, Mills SE,Leong IT, MacDuff E et al . Ewing’s family of tumours of sinonasal tract and maxillary bone. Head Neck Pathol 2011; 5:8-16. [pubmed]

