Case Report

Meckel-Gruber Syndrome
1,Shailaja Shukla1Mona Bargotya 1Deepti Verma 1Sarika Singh2Manisha Kumar*
- 1Departments of Pathology and
- 2Obstetrics & Gynecology Lady Hardinge Medical College, New Delhi.
- Submitted: Friday, January 10, 2014
- Accepted: Saturday, April 26, 2014
- Published: Tuesday, April 29, 2014
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited
Abstract
Meckel Gruber Syndrome (MKS) is a rare autosomal recessive malformation syndrome characterized by multiple congenital anomalies ultimately leading to the death of fetus in utero or shortly after birth. It is characterized by classical triad of occipital encephalocele, infantile polycystic kidneys and postaxial polydactyly. Diagnosis of MKS is made on the basis of ultrasonographic, clinical and morphological findings. Herein we describe two cases of MKS presenting
as classical triad.
Key words
Meckel Gruber Syndrome, Fetal anomaly, prenatal diagnosis
Introduction
Meckel Gruber Syndrome (MKS) is a rare autosomal recessive malformation syndrome with a neural tube defect leading to death of the fetus in utero or shortly after birth.First reports of MKS were published in 1822 by Johann Friedrich Meckel [1]. G.B.Gruber also published reports of pateints with MKS in 1934 and gave it the name dysencephalia splanchnocystica [2].
MKS is characterized by triad of large polycystic kidneys(100%),occipital
encephalocele(90%),and ostaxial polydactyly(83.3%) [3]. Associated abnormalities include oral clefting, genital anomalies, CNS malformations and liver fibrosis.Mortality rate is 100% with most fetuses surviving only few days to weeks. Pulmonary hypoplasia is the leading cause of death. Worldwide incidence is 1/13,250-140,000 live births.There is a predilection for Belgian 1/3000) and Finish (1/9000) populations [4]. In India, highest incidence is in Gujrati Indians (1 affected birth per 1,300) [5].
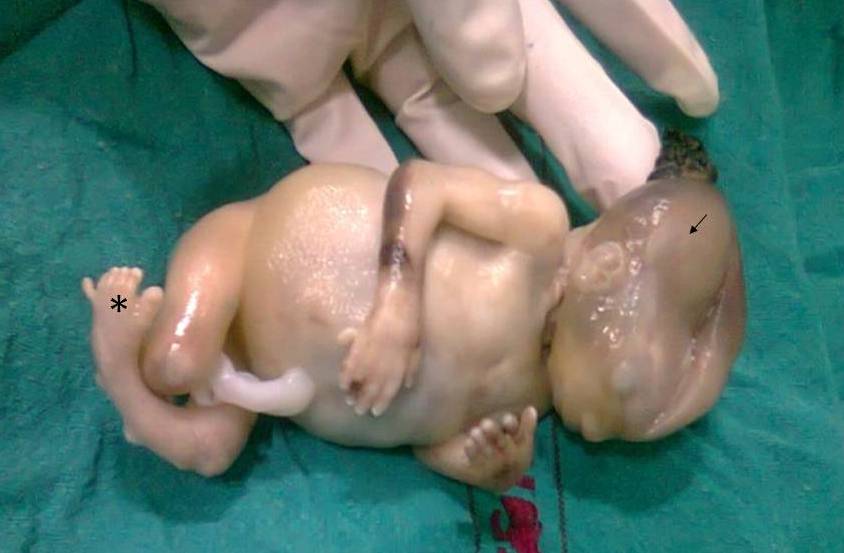
Figure 1: Case I showing encephalocele (à)
and polydactyly (*)
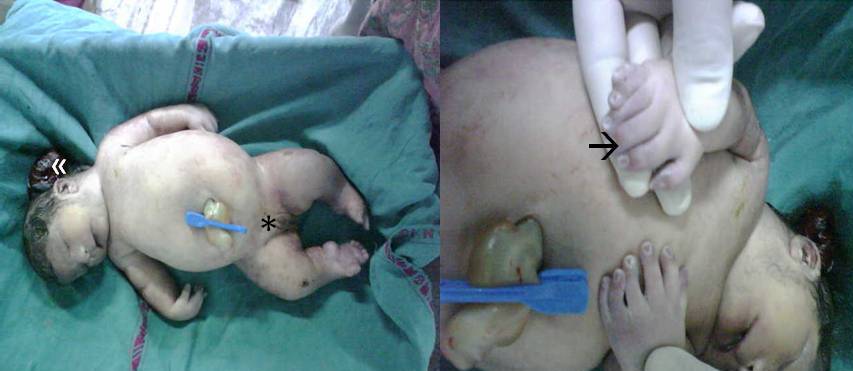
Figure 2: Case II showing encephalocele («), ambiguous genitilia(*) and
polydactyly (à)
Case Presentation
The two cases discussed here were fetuses with abnormal prenatal findings detected during routine ultrasonographic examination. The salient features of both cases are summarized in [Table 1]
(Figure 1 and 2).
TABLE 1: Clinical details of the cases
|
Age of mother/
gestational age
|
Consanguinity
|
Sex of fetus
|
MKS diagnosed at
|
Outcome of
pregnancy
|
Clinical findings
|
|
Case
I
(Fig 1 )
|
20 yrs/16 wks
|
1st
degree
|
male
|
11 wks
|
Dilatation and
Currettage
|
16 wks,
encephalocele,polydactyly bilateral cystic kidneys, oligohydramnios
|
|
Case
II
(Fig 2)
|
24 yrs/36 wks
|
1st
degree
|
male
|
36 wks
|
Still born breech
delivery
|
36 wks,
encephalocele,polydactyly bilateral cystic kidneys,ambiguous
genitilia,oligohydramnios
|
Histopathological Findings
Grossly, kidneys in both the cases were enlarged showing
multiple cysts and loss of corticomedullary differentiation. Liver showed two
cysts measuring 0.2 and 0.8 cm in diameter in case II and autolytic changes in
case I (Figure 3) and (Figure 4).
However, no cysts in liver were identified in Case I. microscopically, sections
from kidneys showed multiple thin walled cysts throughout the parenchyma lined
by low cuboidal epithelium and separated by loose connective tissue. Very few
glomeruli were seen compressed at the periphery (Figure 5).
Sections from liver showed features of ductal plate malformation:proliferation
and dilatation of the bile ducts arranged in a ring like manner in few portal
tracts with increase of collagenous connective tissue i.e.portal fibrosis
(Figure 6). Extramedullary hematopoeisis was also noted.Macroscopic cysts in Case II were lined by columnar épithélium.
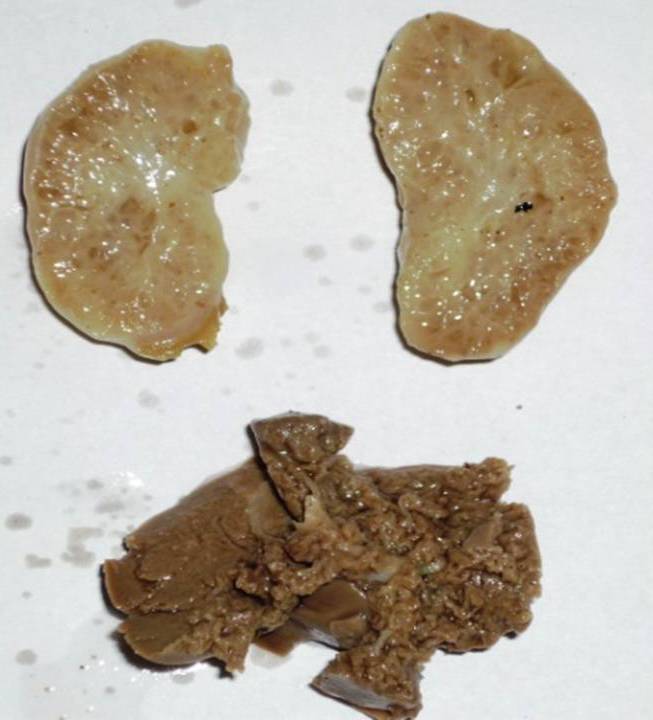
Figure 3: Bilateral enlarged kidneys showing multiple cysts and liver
showing autolytic changes in Case I
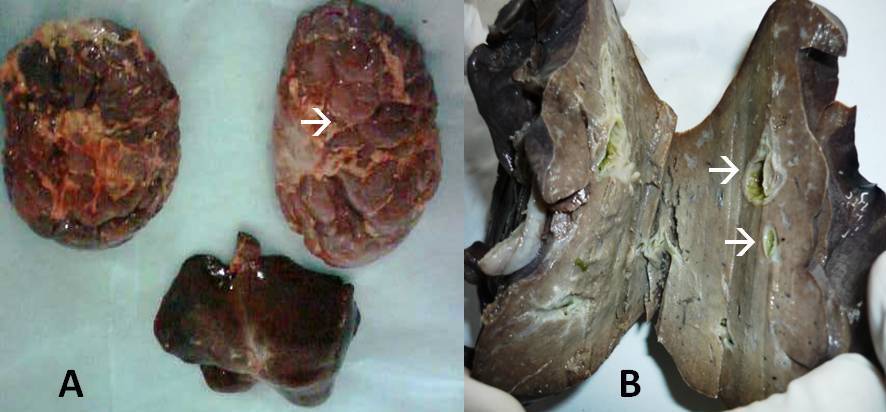
Figure 4:
Gross specimen (A):Bilateral enlarged kidneys showing multiple cysts(à)
in Case II, (B): Liver showing cysts (à)
in case II
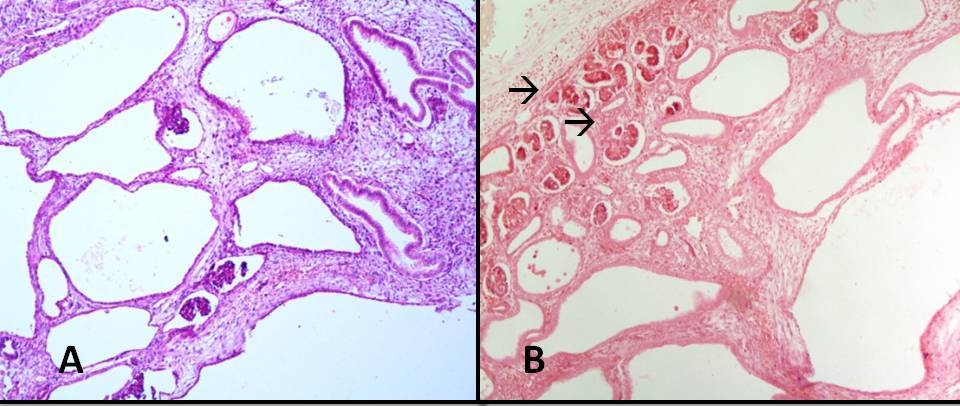
Figure 5:
Photomicrograph showing (A) Multiple renal cysts lined by low low cuboidal
epithelium in case I (H&E x100) (B): Multiple renal cysts with
few glomeuli (à)
in case II (H&Ex100)
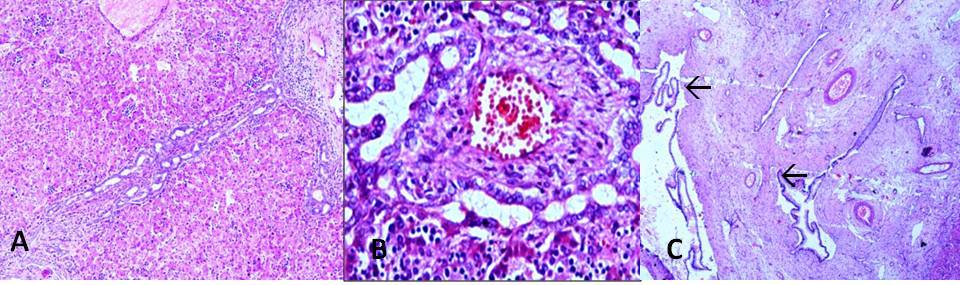
Figure 6:
Photomicrograph showing (A): Ductal plate malformation of liver in case I with
partial autolytic changes (H&Ex100) (B):
Liver showing proliferation of bile ducts
in a ring like manner in case II
(H&Ex400) (C):Liver cyst lined by
columnar epithelium(à)
in Case II(H&E x100).
Discussion
Significant advances have been made in the clinical diagnosis, understanding the pathogenesis and management of MKS. Multiple genetic mutations have been mapped to 6 different loci in chromosomes17q21-24(MKS1),11q13(MKS2),8q21.3q22.1(MKS3)[6],12q21.31q21.33(MKS4)[7],16q12.2(MKS5)[8] & 4p15.3(MKS6) [9].
In the present cases, there was no history of similar fetal anomalies in the siblings as the mother of case I was a primigravida and case II had a live and healthy two year old sibling. No genetic studies could be done as the facility is not available in our institute.
It is important to thoroughly examine any pregnant female with past history of fetal anomalies in previous pregnancies as there is high risk of recurrence (25%). Early (11-14th week) prenatal ultrasonography is the best method to diagnose MKS [10,11]. Subsequent autopsy and chromosomal or molecular studies are confirmatory.
Key message
MKS is a lethal disorder with 100% mortality. An affected infant even if born alive dies shortly thereafter. Termination of pregnancy at risk of MKS can be offered if detected early by prenatal ultrasonography
Authors' Contribution
SS: Pathological diagnosis and edited final manuscript.
MB: Literature search and drafted manuscript.
DV: Literature search and helped in interpretation.
SS: Helped in drafting manuscript.
MK: Treated the patients and provided clinical details.
Conflict of Interests
The authors declare that there is no conflict of interests
Ethical Considerations
Written informed consent was obtained from the mother of the patient for publication of this case report.
Funding
None declared
Acknowledgement
None declared
References
[1]Meckel JF. Beschreibung zweier, durch sehr ahnliche bildungsabweichungen entstelter geschwister. Dtsch Arch Physiol 1822;7:99-172.
[2]Gruber BG. Beitrage zur frage "gekoppelter" missbildungen. (Acrocephalo-Syndactylie und Dysencephalia splanchnocystica). Beitr Path Anat 1934;93:459-76.
[3]Sergi C, Adam S, Kahl P, Otto HF. Study of the malformation of the ductal plate of the liver in Meckel syndrome and review of other syndromes presenting with this anomaly. Pediatr Dev Pathol. 2000;3:568–583[pubmed]
[4]Salonen R, Norio R. The Meckel syndrome in Finland: epidemiologic and genetic aspects. Am J Med Genet. 1984;18:691–698[pubmed]
[5]Young ID, Rickett AB, Clarke M. High incidence of Meckel syndrome in Gujarati Indians.IMed Genet 1985;22:301-4[pubmed]
[6]Smith UM, Consugar M, Tee LJ, McKee BM, Maina EN, Whelan S. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat Genet. Feb 2006;38(2):191-6.[pubmed]
[7]Baala L, Audollent S, Martinovic J, Ozilou C, Babron MC, Sivanandamoorthy S. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am J Hum Genet. Jul 2007;81(1):170-9.[pubmed]
[8]Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. Jul 2007;39(7):875-81[pubmed]
[9]Tallila J, Jakkula E, Peltonen L, Salonen R, Kestila M. Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliopathy puzzle. Am J Hum Genet. Jun 2008;82(6):1361-7[pubmed]
[10].Sepulveda W, Sebire NJ, Souka A, Snijders RJ, Nicolaides KH. Diagnosis of the Meckel-Gruber syndrome at eleven to fourteen weeks’ gestation. Am J Obstet Gynecol. 1997;176:316–319[pubmed]
[11].Mittermayer C, Lee A, Brugger PC. Prenatal diagnosis of the Meckel-Gruber syndrome from 11th to 20th gestational week. Ultraschall Med. 2004;25:275– 279. [pubmed]

