Case Report

Non – Functioning Adrenocortical Carcinoma – Report of a Case and Review of Literature
Nagarekha Kulkarni
Professor, Department of Pathology, Vijayanagara Institute of Medical Sciences, Ballari– 583103, Karnataka, INDIA
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Submitted: January 23, 2017
Accepted: March 31, 2017
Published: April 11, 2017
Abstract
Introduction
Adrenocortical carcinomas (ACC) are rare aggressive tumors with poor prognosis. Their annual incidence is 0.6 – 1.67 cases per million population per year.
Case Presentation
A 48 yrs old male presented with pain per abdomen and a palpable mass on the left hypochondrium with radiating pain in the left lower limb and left side of scrotum. US, CT abdomen and pelvis revealed well defined heterogeneous mass lesion measuring 14 x 12 cms noted in the left suprarenal region. Adrenal gland was not visualized separately. Left kidney was displaced inferiorly. Hormonal assay, renal function test were normal. Left adrenalectomy was done and histopathologic examination revealed adrenocortical carcinoma – non functional.
Conclusion
ACC is a rare aggressive tumor with poor prognosis. In the present case pressure symptoms was an early manifestation. Any patient presenting with the pressure symptoms should be investigated and treated at the earliest, as early diagnosis increases the chances of long time survival.
Key Words
Left hypochondrium, adrenocortical carcinoma, heterogeneous
Introduction
Adrenocortical carcinomas (ACC) are rare aggressive tumors. Their annual incidence is 0.6 – 1.67 cases per million population per year [1]. Overall these tumors accounts for only 0.2% of the causes of death from cancer [2]. It has a bimodal age distribution which peaks in the first and fifth decades of life. Most ACCs are large and weighs from 0.1 to 1 kg. The prevalence of tumors with a diameter of > 1.5 cms is 1.8% and with a diameter of < 6 cm is 0.025% [3]. About 60% of the tumors are functional and rest are non-functional ACCs.
ACC can be broadly divided into hyperfunctioning (hormone-secreting) and nonfunctioning (non-hormone-secreting). Functioning ACC presents earlier with hormonal manifestations such as virilization, feminization, or Cushing’s syndrome. However, nonfunctioning tumors pose a diagnostic challenge because they are diagnosed incidentally due to a mass effect or metastatic disease. Moreover, successful radical management is seldom achievable because most diagnoses are made when the tumor has either invaded local structures or metastasized, explaining the poor prognosis associated with these masses [4]. Herewith, a rare case of non-functioning ACC in a 48 yrs old male is presented.
Case Report
A 48 years old male presented to the surgery OPD with a pain per-abdomen since 1 year. Patient was apparently alright 1 year back. Pain per abdomen was severe in nature since 3 days, continuous, localized to left side of abdomen radiating to lower limbs and scrotum on the left side. There were no aggravating or relieving factors. Patient had mild fever and complains of loss of weight. No history of vomiting or constipation. No history of burning micturition, frequency or urgency, no straining during urination. History of headache was present but no palpitation or perspiration. Patient takes mixed diet, smoker, and alcoholic. He stopped smoking and taking alcohol since one month. Treatment history & family history was insignificant. On general physical examination – patient was conscious, co-operative and well oriented. Per abdominal examination revealed the shape was scaphoid. All quadrants were moving equally with respiration. Flanks were full on left side and veins were not dilated. On palpation abdomen was soft, tender, no guarding rigidity was present. Mass was palpable measuring 10 x 7 cms in the left hypochondrium. The mass was smooth, firm, lower border was smooth and upper border cannot be made out. Fingers can be inserted in the coastal margin. External genital examination revealed no meatal stenosis, left side varicoele was present - grade III. Bilateral testes were palpable. There were dilated veins on the left leg. Cardiovascular system and respiratory system were normal. Vital signs were normal. Clinically, a diagnosis of retroperitoneal mass was made. Patient was subjected for laboratory investigations. Blood investigations, liver function tests and renal function tests were within normal limits. Urine complete and microscopic examination was normal. Urine cortisol levels and urinary vanillyl mandelic acid were normal. Fasting blood sugar was 70mg/dl as shown in
table -1.
Table 1: Showing laboratory findings
|
Parameter
|
Observed Value
|
Normal Range
|
|
Hb%
|
8 g/dl
|
(12.5-14.5g/dl)
|
|
RBC Count
|
3.3 millions/cmm
|
(4.5-5.5millions/cmm)
|
|
Haematocrit
|
25 %
|
(35% to 45%)
|
|
WBC Count
|
6,300 cells/cmm
|
(4000 to 11,000/cmm)
|
|
Differential Count
|
Neutrophils: 81 %
|
|
|
|
Lymphocytes: 19 %
|
|
|
|
Eosinophils: 00%
|
|
|
|
Monocytes: 00%
|
|
|
|
Basophils: 00%
|
|
|
Platelet Count
|
2.3 lakhs/cmm
|
|
|
Peripheral smear
|
Normocytic normochromic anemia
|
|
|
Random Blood Sugar
|
140 mg/dl
|
(100 - 140 mg/dl)
|
|
Blood Urea
|
19mg/dl
|
(5 to 25mg/dl)
|
|
Serum Creatinine
|
0.8mg/dl
|
(0.8 to 1.2mg/dl)
|
|
Serum sodium
|
150meq/lt
|
(135-150 meq/lt)
|
|
Serum potassium
|
3.5 meq/lt
|
( 3.5-5.5 meq/lt)
|
|
Serum chloride
|
100meq/lt
|
(98-115meq/lt)
|
|
Urine cortisol levels
|
318µ/24 hrs
|
(28.50 to 213µ/24hrs)(CLIA)
|
|
Urine Vanillyl mandlic acid
|
8.3 g/24hrs
|
(< 13.6mg /24hrs) (column
chromatography)
|
|
Testosterone
|
659ng/ml
|
(28 to 11 ng/ml) (CMIA – ABBOTT)
|
Patient was then subjected for Ultra- sound (US) and Computed tomography (CT) abdomen & pelvis which revealed well defined heterogeneous mass lesion measuring 14 x 12 cms noted in the left suprarenal region. Adrenal gland was not visualized separately. Left kidney was displaced inferiorly. All the other organs were normal. The metastatic work-up included CT scans of the head & chest, magnetic resonance imaging (MRI), and a bone scan. The head and chest CT scan revealed normal findings. Neither the bone scan nor MRI showed evidence of bone metastasis. Guided FNAC was not done, because surgery was done on emergency basis. Patient underwent left adrenalectomy, as the adhesion were more, splenectomy was done. The adrenalectomy mass and spleen was sent for histopathological examination.
Both the specimens were received in the histopathology section. Macroscopically, the adrenalectomy mass was large, solid, grey white to grey brown, globular, capsulated mass of tissue measuring 16X 14 X 12 cms and weighing 1600 gms
(Figure 1). External surface was congested. On cut section variegated grey brown areas with soft friable intra - tumoral nodules were seen. In the centre yellowish areas with focal areas of hemorrhage and necrosis were seen.
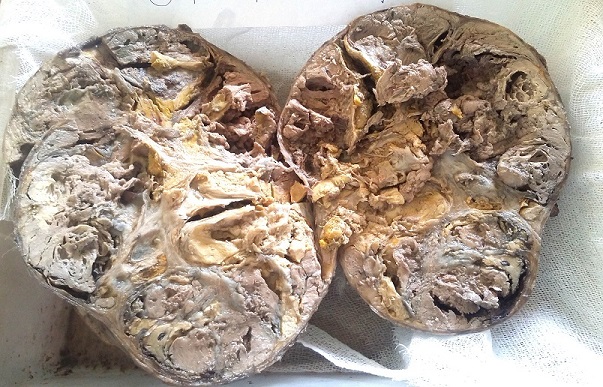
Figure 1: Cut section of the
adrenalectomy specimen showing
variegated grey brown
areas with soft friable intra tumoral nodules associated with hemorrhage and
necrosis
Microscopically, multiple sections from solid, capsulated mass showed tumor cells which was arranged predominantly in a trabecular pattern, also seen were ribbon like and small nodular pattern separated by fine fibrocollagenous septae. The tumor cells were pleomorphic with abundant granular and vacuolated eosinophilic cytoplasm, large pleomorphic, vesicular nuclei and prominent nucleoli. The mitotic activity was 10-12/50 HPF
(Figure 2, 3). At few areas atypical mitosis, binucleated and multinucleated tumor giant cells were seen. There was vascular invasion and capsular invasion. Large areas of hemorrhage and necrosis were seen. A diagnosis of adrenocortical carcinoma was made. The spleen macroscopically and microscopically was normal. No pathologic diagnostic findings were present.
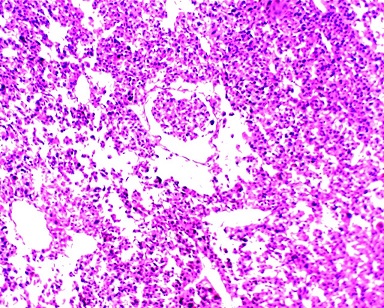
Figure 2: Tumor cells are arranged in trabeculae and sheets
with large pleomorphic vesicular nuclei and prominent nucleoli with vascular
invasion (H/E – 10X)
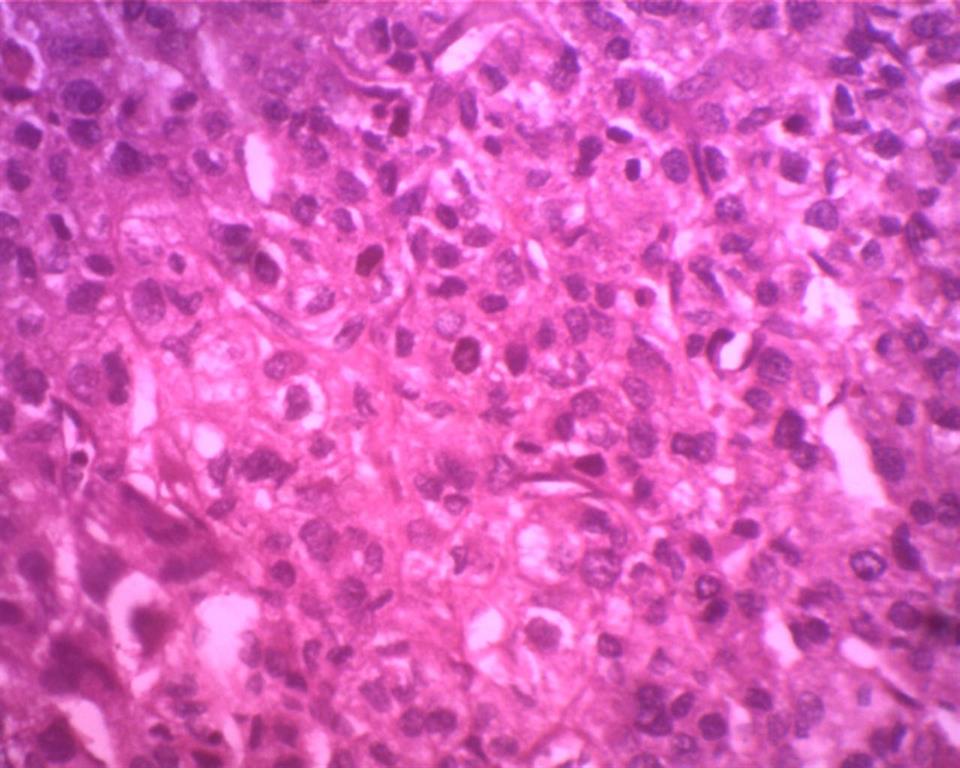
Figure 3: Tumor cells are arranged in trabeculae and sheets
with large pleomorphic vesicular nuclei and prominent nucleoli with mitosis
(H/E- 40X)
After considering laboratory, radiological and histopathological findings a diagnosis of –non-functioning adrenocortical carcinoma was made.
Discussion
Adrenal cortex tumors are rare. The most common tumor among them are ACC and accounts for 6% of adrenal tumors. ACC is a rare, progressive heterogeneous tumor with poor prognosis. Among all the cancers, ACC represents 0.02% [2]. These tumors are common in the first and fifth decade of life. They are classified into functional and non- functional. The non-functional ACCs are asymptomatic and are diagnosed based on clinical evaluation, hormonal assays, radiological and histopathological features. They remain undiagnosed till late and present with a large mass and metastatic disease. The incidence of non-functional ACCs is less common. They occur in elderly age group (> 30 yrs) and are more common in men (male to female ratio is 3:2) [5]. The above mentioned data is similar to the present case. However, Wooten et al and Alloho et al studies showed non-functional ACCs were more common in females (58.6%) than males (41.1%) [6,
7].
The presenting symptom in the present study was pain in the left side of the abdomen which was radiating to the left lower limb and left scrotum. Later on it was diagnosed radiologically as left varicocele. Patient also complained of loss of weight. Also these findings were similar to Cheungpasitporn et al studies [8]. But few cases were diagnosed incidentally or vague abdominal pain and loss of weight [7,
9].
Imaging studies using US, CT, MRI and more recently positron emission tomography (PET-FDG) to observe adrenal mass size and appearance have been used to distinguish between benign and malignant lesions. None of these imaging techniques is specific for adrenal carcinoma but each modality is complementary to the others. On US, smaller lesions are homogenous, whereas larger lesions are heterogeneous secondary to necrosis/hemorrhage. CT is the next imaging modality used. Adrenal carcinomas tend to be large (>6 cm) irregularly shaped mass with central areas of necrosis and hemorrhage, resulting in variable enhancement. Calcification is seen in up to 30% of cases.
If the mass is small, then it is difficult to distinguish from an adenoma, as aggressive features are often absent. If CT is inconclusive MRI can be useful. MRI shows heterogeneous mass as high signal on T2 sequences. MRI sensitivity for benign and malignant tumor differentiation varies from 81-89% and specificity is from 92-99%. A useful characteristic of MRI is that it can identify tumor invasion in the inferior vena cava (IVC) enabling better surgical planning [9]. Ninety two percent of ACCs are large tumors of > 6 cms in diameter. These tumors also efface the native adrenal gland as in the present case. But in P Kaur
et al., [10] case on CT the size was 3 x 4 cms which was locally invading and diagnosed incidentally. Incidentaloma may be primarily adrenal tumors, metastatic malignancies or non-neoplastic lesions. The differential diagnosis of these adrenal masses include: cortical adenoma, pheochromocytoma, aldosteronomas, metastatic lesions, adrenal cortical carcinomas, and a host of non –neoplastic lesions. To differentiae one another further investigations are necessary.
The laboratory findings in non-functional ACCs are normal except hypoglycemia. Because non–functional ACCs are associated by a deletion in some enzyme required for cortisol synthesis. In the present study all the laboratory findings were normal and fasting blood sugar was 70mg/dl. The BP and urine vanillyl mandlic acid level was normal which ruled out pheochromocytoma. Renal functional tests were normal; there was no hematuria or any problem with urination which again ruled out renal cell carcinoma. The efficacy of guided FNAC in the diagnosis of adrenal lesions was seen in many cases. It distinguishes between adrenal tumor from metastasis. But it cannot distinguish between adenoma from carcinoma. In the present case FNAC was not done. Keeping in view the radiological and laboratory findings adrenalectomy was done.
In the present case macroscopically, adrenal mass was large, well-encapsulated. On cut section areas of haemorrhage and necrosis were seen. This again indicates malignancy.
Diagnosing microscopically, with capsular or vascular invasion was the reliable sign of cancer. In the absence of these findings Weiss histopathological scoring system can be used. This method is simple and most reliable for assessing the malignancy. The neoplasm was classified according to histologic criteria of Weiss. Features associated with and increased probability of a malignant clinical behavior included tumor weight (> 400 g), tumor size (> 10.5 cm), vena cava invasion, severe nuclear atypia, > 5 mitotic /50 high power fields and the presence of atypical mitotic figures, nuclear grade III or IV, clear cell comprising 25% or less of the tumor, a diffuse architecture, microscopic necrosis and invasion of venous, sinusoidal and capsular structures. Previously, the presence of 4 or more of these histologic findings was defined as indicative of malignancy, later it was modified to 3 or more [11]. In the present case four criteria were present; high mitotic figures, capsular invasion, necrosis, a diffuse architecture. A score higher than 3 is suggestive of malignancy. The present case met the above mentioned criteria.
In majority of the cases, the tumor has either invaded adjacent organs or already metastasized to distant organs at the time of intial diagnosis [12]. In the present case, there was no local invasion of adjacent organs and no distant metastases. As per the Fassnacht M
et al., modified staging system ACCs were staged as follows: T1:tumor < 5cms, T2: tumor > 5cms, T3: tumor infiltration of surrounding tissues, T4: tumor invasion of adjacent organs, vena cava or renal vein, N0; no nodes, N1: positive lymph nodes, M0: no metastasis, M1: distant metastasis. Therefore stage 1: T1N0M0, stage 2: T2N0M0, stage 3: T1-2, N1, M0 or T3-4, N0-1, M0 and stage 4 : any T, any N, and any M1 [13]. The present case belongs to stage 2 – T2N0M0.
These tumors shows positive for immunohistochemistry markers like vimentin, MelanA, synaptophysin, Neurofilament(NF) and α-inhibin overexpression. Negative for cytokeratin, chromogranin A [14]. In the present case IHC marker study was not done because the facility was not available.
The treatment of choice is complete excision, if needed surrounding structures can be removed. In the present case complete excision with splenectomy was done and patient is under follow up. Radical surgical resection of adrenocortical carcinoma offers the best chance for prolonged recurrence free survival. Recurrence of adrenal cortical carcinoma after radical surgery is a common finding although successful reoperations have been reported with encouraging results [12]. Unresectable or widely disseminated tumors may be palliated by antihormonal therapy with mitotane, systemic chemotherapy, or (for localized lesions) radiation therapy. Although the survival for patients with stage IV tumors is usually less than 9 months. Palliative chemotherapy with cisplatin-based regimens has produced objective responses in approximately 30% of patients treated [14,
15]. Frequent follow up visits and examinations are necessary after adrenalectomy. Only early diagnosis of relapse gives the chance of a successful re-operation. After adrenalectomy the follow-up abdominal and chest CT should be performed every 3–4 months for the first two years. Later the CT exams can be performed less often. Regardless of the adrenocortical cancer treatment, prognoses for patients remain poor. But like in other malignant neoplasm cases early diagnosis increases the chances of long time survival.
Conclusions
ACC is a rare aggressive tumor with poor prognosis. In the present case pressure symptoms was an early manifestation. Radiological findings and histopathological examination was helpful in diagnosing the present case. There was no evidence of metastasis. Surgical excision was the treatment of choice. An aggressive strategy for recurrent and metastatic ACCs is advisable. If the patient presents in an early stage and undergoes surgical excision the survival rate is increased.
Author's Contribution
Did the literature search prepared and edited the manuscript
Ethical Considerations
Written informed consent was taken from the patient, a copy of consent is available with authors
Conflict of Interests
The author declare that there is no conflict of interest.
References
1. Zografos GC, Driscoll DL, Karakousis CP, Huben RP. Adrenal carcinoma: review of 53 cases. J Surg Oncol 1994; 55:160-164.
[Pubmed]
2. Favia G, Lumachi F, Carraro P, D'Amico
DF. Adrenal cortical carcinoma. Our experience. Minerva Endocrinol 1995; 20(1): 95-9.
[Pubmed]
3. Russi S, Blumenthal HT, Gray SH. Small adenomas of the adrenal cortex in hypertension and diabetes. Arch Intern Med, 1945. 76: p. 284-291
[Pubmed]
4. Luton JP, Cerdas S, Billaud L, Thomas
G, Guilhaume B, Bertagna X, Laudat MH, Louvel A, Chapuis Y, Blondeau P,et al: Clinical features of adrenocortical carcinoma, prognostic factors, and the effect of mitotane therapy. N Engl J Med 1990; 322(17): 1195-201.
[Pubmed]
5. Kishikawa H, Mizuno T, Takagi I, Yamakawa Y, Shimozato T,
Honda K, Tatematsu M. Nonfunctioning adrenocortical carcinoma in a young girl. Jpn J Surg. 1985; 15: 477-82.
[Pubmed]
6. Wooten MD, King DK: Adrenal cortical carcinoma. Epidemiology and treatment with mitotane and a review of the literature .Cancer 1993; 72(11): 3145-55.
[Pubmed]
[Full
text]
7. Allolio B, Fassnacht M. Clinical review: Adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab. 2006; 91: 2027-37.
[Pubmed]
8. Cheungpasitporn W, Horne JM, Howarth CB. Adrenocortical carcinoma presenting as varicocele and renal vein thrombosis: a case report. J Med Case Reports. 2011; 5: 337.
[Pubmed]
[PMC
Full text]
9. Barzon L, Boscaro M. Diagnosis and management of adrenal incidentalomas. J Urol 2000; 163: 398-407.
[Pubmed]
10. Kaur P, Chauhan A, Singh G, Kataria S, Mathur S. Incidentally discovered nonfunctioning adrenal cortical carcinoma: a case report and review of literature. The Internet Journal of Third World Medicine. 2008; 7: 2.
[Free
Full text]
11 Lau SK, Weiss LM. The Weiss system for evaluating adrenocortical neoplasms: 25 years later. Hum Pathol. 2009; 40: 757-768.
[Pubmed]
12. Agrons GA, Lonergan GJ, Dickey GE, Perez-Monte JE. Adrenocortical neoplasms in children: radiologic-pathologic correlation. Radiographics. 1999; 19(4): 989-1008.
[Pubmed]
13. Fassnacht M, Johanssen S, Quinkler M,
Bucsky P, Willenberg HS, Beuschlein F, Terzolo M, Mueller HH, Hahner S, Allolio
B; German Adrenocortical Carcinoma Registry Group; European Network for the
Study of Adrenal Tumors. Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: proposal for a Revised TNM Classification. Cancer. 2009; 115(2): 243-50.
[Pubmed]
[Free
Full text]
14 Hesketh PJ,McCaffrey RP. Finkel HE,
Larmon SS, Griffing GT, Melby JC. Cisplatin based treatment of adrenocortical carcinoma. Cancer Treat Rep 1987; 71(2): 222-4.
[Pubmed]
15. Tattersall MH, Lander H, Bain B,
Stocks AE, Woods RL, Fox RM, Byrne E, Trotten JR, Roos I. Cis- plantinum treatment of metastatic adrenal carcinoma. Med J Aust 1980; 1(9):419-21.
[Pubmed]

