Case Report

Primary Histopathologic and Immunohistochemical Diagnosis of Malignant Pheochromocytoma of Adrenal gland-Case report and Review of Literature
*,Hema Aiyer *Toshima Kukreja *Gaurav Sharma
- * Anatomic Pathology, Dr Lal Path Labs, New Delhi-110085, India
- Submitted Tuesday, April 29, 2014
- Accepted:Thursday, May 22, 2014
- Published Thursday, September 04, 2014
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited
Abstract
Malignant pheochromocytomas (PCC) are rare tumours. In the absence of distant metastasis, diagnosis of malignancy on histological grounds has always been challenging and controversial. We report a case of a 32 Yrs. female who presented with abdominal pain and was incidentally diagnosed as having right adrenal mass. Based on histology (using Pheochromocytoma Adrenal Gland Scaled Score – PASS) and IHC a diagnosis of Pheochromocytoma with features of malignancy was given. Patient underwent excision preceded by NACT and is doing fine till date.
Key words
Malignant pheochromocytoma, PASS score.
Introduction
Malignant pheochromocytomas / paragangliomas are rare tumours (10% of all pheochromocytomas) [1] with a poor prognosis. The pathological diagnosis of malignancy in pheochromocytomas remains a controversial issue [2].
The morphologic differentiation of benign and malignant PCCs is difficult [3]. The presence of metastatic lesions and gross invasion into surrounding tissues are the only acceptable parameters to confirm malignancy [4] with the most common metastatic sites being local lymph nodes, Bone (50%), Liver (50%) & Lung.[Table 1]
Table.1
|
Parameters
|
Score
|
Present Case
|
|
Vascular invasion
|
1
|
0
|
|
Capsular invasion
|
1
|
0
|
|
Periadrenal adipose tissue invasion
|
1
|
0
|
|
Large nests / diffuse growth
|
2
|
0
|
|
Focal / confluent necrosis
|
2
|
2
|
|
High cellularity
|
2
|
2
|
|
Tumour cell spindling
|
2
|
2
|
|
Cellular monotony
|
2
|
0
|
|
Increased mitosis >/10HPF
|
2
|
2
|
|
Atypical mitosis
|
2
|
0
|
|
Profound nuclear
pleomorphism
|
1
|
1
|
|
Hyperchromasia
|
1
|
1
|
Studies on malignant pheochromocytomas have been inconclusive and anecdotal due to the low incidence of the tumour and limited post operative follow up with inconsistent protocols.
We discuss a case of malignant pheochromocytoma that occurred in a 32 Yr female. On initial needle biopsy a possibility of cortical carcinoma was entertained on histology. IHC performed on the initial core biopsy showed features suggestive of pheochromocytoma and an adrenalectomy was performed.
Case History
A 32 Yr old female presented with central abdominal pain off & on for 1 month. She had a history of abdominal hysterectomy done 2 years back. On examination, scar of previous hysterectomy incision was present. On USG abdomen a right adrenal mass was incidentally discovered with contracted thick walled gall bladder and cholelithiasis. The mass was 11.0 x 10.0 x 8.0 mm in size. The possibilities suggested were:
Carcinoma, gallbladder with right adrenal metastasis &
Pheochromocytoma.
All hematologic parameters and biochemical serum tumour markers (VMA) were within the normal range. Based on the core needle biopsy findings, the patient received 1 cycle of Neoadjuvant Chemotherapy (Gemcitabine& Benzoxin) with an ECOG performance status of 1. The Chemotherapy was given based on the histologic diagnosis of poorly differentiated carcinoma / cortical carcinoma prior to IHC Therefore it was tailored to suit a cortical carcinoma
The initial core needle biopsy from the right adrenal mass was measuring 0.3 x 0.2 x 0.1 cm and a diagnosis of poorly differentiated carcinoma with areas of necrosis, possibly an Adrenal cortical carcinoma was suggested. No mature adrenal parenchyma was identified and IHC was advised for definite evaluation.
IHC on trucut biopsy showed positivity for Chromogranin A. CK20 Calretinin and inhibin were negative. CK7 showed patchy immunoreactivity IHC findings were consistent with a Pheochromocytoma. Post Neoadjauvant Chemotherapy the right adrenalectomy specimen was received 3 months later in multiple grey brown friable tissue pieces together measuring 11 x 7 x 5 cm and weighing 160 gm in aggregate. Cut surface of the mass was grey white friable and hemorrhagic. No normal adrenal was identifiable grossly. On microscopic examination the tumour was composed of large polygonal cells arranged in sheets with extensive necrosis (figure 1A) and pleomorphic large nuclei with focal spindling (figure 1B). The mitotic rate was >3/10HPF (10/10HPF). A repeat Immunohistochemistry was run on the resection specimen. The tumour cells were Immunoreactive for Chromogranin (figure 1C). Synaptophysin (figure 1D) and focally for CK. Inhibin, Calretinin & CK7 and CK20 were – Non-immunoreactive. The Ki-67 labelling Index was >4% (figure 1E) S-100 was Non-immunoreactive (figure 1F)
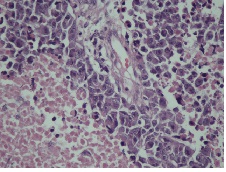
Figure 1A: Polygonal tumor cells arranged in sheets with extensive necrosis
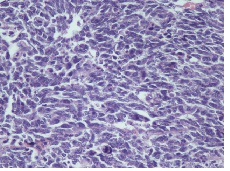
Figure 1B: Tumor cells with pleomorphic large nuclei and focal spindling
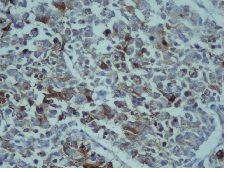
Figure 1C: Tumor cells Immunoreactive for Chromogranin-A
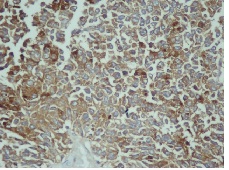
Figure 1D: Tumor cells Immunoreactive for Synaptophysin
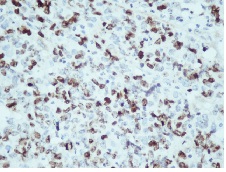
Figure 1E: Ki-67 Immunostaining showing a Labelling index >4%
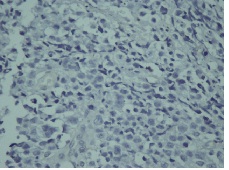
Figure 1F: Negative immunostaining for S-100
The tumour was scored according to the Pheochromocytoma of Adrenal Gland Scaled Score (PASS) system as under:-Based on PASS scoring system our case scored 12 (PASS score> 4 shows aggressive behaviour and a PASS score <4 behave in a benign fashion).
Thus based on morphology (PASS score) and IHC a diagnosis of Pheochromocytoma with features suggestive of malignancy was given.
Discussion
Pheochromocytoma is an infrequent tumour, originating from the adrenal medulla. The triad of headache, sweating and palpitations in patients with hypertension is diagnostic; with 99% specificity & 91% sensitivity, for Pheochromocytoma. There is considerable controversy as to whether the histopathological appearance of chromaffin cell tumours can predict malignancy in the absence of distant metastasis [5]. According to the WHO, malignancy is defined by the presence of metastasis. Histologic characteristics have not proved helpful for diagnosis of malignancy and for prediction of prognosis [6]. Additional markers that might be useful include immunohistochemical expression of MIB-1, Bcl-2, Cathepsin B & D, βFGF & Type IV collagenase of which MIB-1 labelling of >3% has yielded a specificity of a 100% and sensitivity of 50% in [7 8] predicting malignancy.
Wailly P et al compared the size, weight, Ki-67 index and necrosis in 53 proven cases of malignant pheochromocytoma with PASS score. They concluded that size and weight of pheochromocytoma directly correlated to PASS and malignancy. Presence of necrosis, Ki-67 index >4% and pS-100 absence suggested a high risk of malignancy recurrence [9 10].
Pheochromocytoma has good overall prognosis with a 5 year survival of more than 95% in benign tumours and recurrences below 10%. For malignant tumours due to their low incidence it is difficult to determine their outcome.
Thus to conclude in an a symptomatic patient the incidental discovery of Pheochromocytoma with features suggestive of malignancy on the basis of histology and IHC can guide the oncologist towards further management, Chemotherapy and follow up.
Authors' Contribution
HA- Conceived the report and edited the final manuscript
TK-Carried out the literature search and prepared the manuscript
GS-Helped in the design and drafting of the manuscript
Conflict of Interests
The authors declare that there are no conflict of interests.
Ethical Considerations
Written consent was obtained from the patient for publication of this case
report
Funding
None
Acknowledgement
None
References
[1].Adler JT, Meyer – Rochow GV, Chen H, Benn DE, Robinson BG, Sippel RS, Sidhu SB. Pheochromocytoma: Current approaches and Future Directions. The Oncologist 2008; 13:779-93[Pubmed]
[2].Chrisoulidou A, Katsas G, Ilias I, Grossman AB. The diagnosis and management of Malignant Pheochromocytoma and Paraganglioma. Endocr Relat Cancer 2007; 14:569-85.[Pubmed]
[3].Eisenhofer G, Bornstein SR, Brouwers FM, Cheung NK, Dahia PL, de Krijger RR, Giordano TJ, Greene LA, Goldstein DS, Lehnert H, Manger WM, Maris JM, Neumann HP, Pacak K, Shulkin BL, Smith DI, Tischler AS, Young WF Jr. Maligant Pheochromocytoma: Current status and initiatives for future progress. Endocr Relat Cancer 2004; 11(3):423-36.[Pubmed]
[4].Scholz T, Eisenhofer G, Pacak K, Dralle H, Lehnert H. Current Treatment of Malignant Pheochromocytoma. J Clin endocrional Metab 2007; 92(4): 1217-25.[Pubmed]
[5].Schlumberger M, Gicquel C, Lumbroso J, Tenenbaum F, Comoy E, Bosq J, Fonseca E, Ghillani PP, Aubert B, Travagli JP, et al. Malignant Pheochromocytoma: Clinical, Biological, Histologic and therapeutic data in a series of 20 patients with distant metastasis. J Endocrinol Invest 1992; 15:631-42.
[6].Huang KH, Chung SD, Chen SC, Chueh SC, Pu YS, Lai MK, Lin WC. Clinical and Pathological data of 10 malignant pheochromocytomas: Long-term follow up in a single institute. Int J Urol 2007; 14(3):181-5.[Pubmed]
[7].Clarke MR, Weyant RJ, Watson CG, Carty SE. Prognostic markers in pheochromocytoma. Hum Pathol 1998; 29(5):522-6[Pubmed]
[8].Nagura S, Katoh R, Kawaoi A, Kobayashi M, Obara T, Omata K. Immunohistochemical estimations of growth activity to predict biological behaviour of pheochromocytomas. Mod Pathol 1999; 12(12):1107-11 [Pubmed]
[9].de Wailly P, Oragano L, Radé F, Beaulieu A, Arnault V, Levillain P, Kraimps JL. Malignant Pheochromocytoma: new malignancy criteria. Langenbecks Arch Surg 2012; 397(2):239-46 [Pubmed]
[10].Thompson LD. Pheochromocytoma of Adrenal gland scaled score (PASS) to separate benign from malignant neoplasms, a clinicopathologic& immunopheotypic study of 100 cases. Am J Surg Pathol. 2002 May, 26 (5):551-66 [Pubmed]


