Case Report

A Case of Endophytic Retinoblastoma
*Shobhna Jorwekar *Akshay Bhandari *Pratik Gogri, *, Dharmdeep Patel
- *Department of Ophthalmology, Rural Medical College of Pravara Institute of Medical Sciences, Loni.
- Submitted:Submitted April, 04, 2013
- Revision submitted on: February 04, 2014
- Accepted: on Saturday, February 08, 2014
- Published:on Friday, May 16, 2014
This is an Open Access article distributed under the terms of the Creative Commons Attribution License ((http://creativecommons.org/licenses/by/3.0)which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited
Abstract
Background
Retinoblastoma is the most common primary intraocular tumor of childhood. It represents approximately 4% of all pediatric malignancies. Though congenital, it is not diagnosed at birth, and usually manifests between 1-3 years of age.
Case Presentation
A 2 years old male child presented with white reflex in right eye since 1-2 months. USG B-scan (ultrasonography B-scan) of right eye revealed hyper echoic mass on the nasal aspect of posterior segment with multiple foci of calcification. CT-scan orbit demonstrated a contrast-enhancing retrolental mass which showed calcifications. Patient was diagnosed to have retinoblastoma. Enucleation of the eye ball was done. Histopathological report confirmed the diagnosis.
Conclusion
Successful treatment and prevention of spread of retinoblastoma can be possible with early and accurate diagnosis and with timely intervention. In case of atypical presentation, high degree of clinical and radiological suspicion is of paramount importance.
Key words
Retinoblastoma; intraocular tumour; enucleation; leucocoria.
Introduction
Retinoblastoma is a primary malignant intraocular neoplasm that arises from immature retinoblasts within the developing retina [1]. Retinoblastoma arises in one of every 15,000 live births. It affects boys and girls with equal frequency and has no known racial predilection. Approximately one in three to one in four affected children will have a family history of retinoblastoma. It results either from sporadic or heritable mutations in the retinoblastoma gene, (RB-1) located on long arm of chromosome 13 [2]. Tumors result from a deletion of the retinoblastoma gene which codes for a protein that acts as an anti-oncogene or tumor suppressor. Retinoblastoma can be inherited as a familial tumor in which the affected child has a positive family history of retinoblastoma or as a nonfamilial (sporadic) tumor in which the family history is negative for retinoblastoma. Approximately 60–70% of retinoblastoma cases are unilateral, and the remaining 30–40% cases are bilateral.
Case Report
A 2 years old male child presented with white reflex in right eye since 1-2 months which was not associated with any other complaints. There was no history of trauma or eye infection. Child was born full term and healthy. Developmental milestones were adequate for the age. There was no history of similar complaints in the family.
General and systemic examination of the patient was normal. Ocular positions were normal & ocular movements in both eyes were full and free in all directions of gaze. On Ophthalmic examination in right eye white reflex was seen in pupillary region (Figure 1). Pupillary reaction to light was sluggish and patient was not following light. Rest of the anterior segment examination was within normal limit. Ophthalmoscopic examination of right eye after sedating the child revealed whitish cauliflower like growth obscuring the details of the fundus. Anterior segment and fundus findings of the left eye were within normal limit. USG B-scan of right eye revealed hyper echoic mass on the nasal aspect of posterior segment with multiple foci of calcification. Left eye revealed no abnormality. There was no evidence of distant metastasis on USG abdomen with pelvis and chest X-ray. Contrast-Enhanced Computed Tomography (CECT) scan reported, a well defined mildly enhancing lesion in the right eye occupying the entire vitreous cavity with associated surrounding retinal detachment and evidence of intralesional calcification with no extrascleral extension, optic nerve involvement or intracranial metastasis. All the routine blood investigations were within normal limit. On the basis of clinical findings, USG and CT scan findings; the tumor is graded as group E according to ICIR (International Classification for Intraocular Retinoblastoma) and group V according to Reese Ellsworth classification.
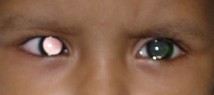
Figure 1: Right eye of patient showing white pupillary reflex (leucocoria)
Patient was posted for right eye enucleation. Specimen was sent for histopathological examination (Figure 2) On histopathological examination, microscopic sections from eyeball showed tumor mass composed of dense masses of uniform small round cells. Individual cells showed hyperchromatic nuclei and scanty cytoplasm (Figure 3) Areas of calcification and necrosis were seen. Sections from optic nerve were free of tumor cells. Clinical diagnosis of retinoblastoma was confirmed. Patient is having regular follow up since then and there is no involvement of other eye till now.
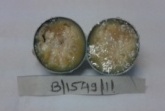
Figure 2: Cut section of the enucleated eye ball
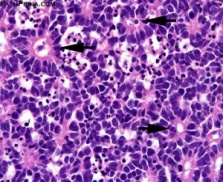
Figure 3: Microscopy showing dense masses of uniform small round cells with hyperchromatic nuclei and scanty cytoplasm showing rosette pattern. (40X magnification)
Discussion
Retinoblastoma arises as a malignant proliferation of the immature retinal neural cells called retinoblasts which are derived from photoreceptor precursor cells. The average age when children are found to have retinoblastoma is 9 to 12 months in bilateral cases and 24 months in unilateral cases [1]. It results from a deletion of the retinoblastoma gene, located on chromosome 13 which codes for a protein that acts as an anti-oncogene or tumor suppressor. Most common presenting manifestation of retinoblastoma is a white pupil (leucocoria), seen in 50-60% of cases. Parents may notice this with direct observation or on photographs. The second most common presenting manifestation is strabismus (20-30%), which may be either esotropia or exotropia [2,3]. Other less common presenting manifestations of retinoblastoma include a red eye, excessive tearing, globe expansion (buphthalmos), corneal clouding, iris neovascularization, clumping of white tumor cells on the iris or in the aqueous humor, spontaneous hyphema, and sterile orbital cellulitis. The most common spread of retinoblastoma occurs through the optic nerve to the brain and invasion of the optic nerve is a poor prognostic sign [4]. Systemic spread of the tumor can occur through lymphatic tissue and blood stream. Retinoblastoma growth patterns are subdivided into intraretinal, endophytic and exophytic. Intraretinal tumors are limited to the substance of the retina. Endophytic retinoblastoma is one that grows from the retina inward toward the vitreous cavity. An exophytic retinoblastoma is one that grows from the retina outward into the subretinal space. Occasionally, a retinoblastoma stops growing spontaneously and loses its malignant character. Such a tumor is called a retinoma.
Grossly retinoblastoma appears chalky white, friable well circumscribed polypoid tumor with dense foci of calcification giving the typical ‘cottage cheese’ appearance. Histopathologically, it consists of small round cells with large nuclei, resembling the cells of the nuclear layer of the retina. Well differentiated tumors include Flexner Wintersteiner rosettes (highly specific of retinoblastoma), Homer-Wright rosettes, pseudo rosettes, fleurettes formation and with areas of necrosis and calcification [5,7]. The diagnosis is easily reached with careful ophthalmologic examination, Ultrasonography, plain x-ray of the orbit and CT scan.
The most important objective in the management of a child with retinoblastoma is survival of the patient and the second most important goal is preservation of the globe. Chemotherapy is the primary therapeutic option in most children with bilateral retinoblastoma. Frequently in bilateral cases, the more severely affected eye is removed. Currently the treatment modalities that we advocate include enucleation, external beam irradiation, brachytherapy, photocoagulation, cryotherapy, chemotherapy, and chemo thermotherapy and chemo reduction [8,9].
Conclusion
Successful treatment and prevention of spread of retinoblastoma can be possible with early and accurate diagnosis with timely intervention. In case of atypical presentation, high degree of clinical and radiological suspicion is of paramount importance.
Authors' Contribution
SJ: conception and design, acquisition, analysis and
interpretation of data, drafting and revising of the article
AB: conception and design, acquisition, analysis and
interpretation of data, drafting and revising of the article
PG: conception and design, acquisition, analysis and
interpretation of data, drafting and revising of the article
DP: conception and design, acquisition, analysis and
interpretation of data, drafting and revising of the article
Conflict of Interests
The authors declare that there are no conflicts of interests.
Ethical Considerations
The written informed consent was obtained from the patient for publication of this case report
Ethical Considerations
The written informed consent was obtained from the patient for publication of this case report
Funding
None
Acknowledgement
None declared
References
[1].Goddard AG, Kingston JE, Hungerford JL. Delay in diagnosis of retinoblastoma: risk factors and treatment outcome. Br J Ophthalmol. 1999; 83(12): 1320-3.[pubmed]
[2].Nork TM, Schwartz TL, Doshi HM, Millecchia LL. Retinoblastoma: Cell of origin. Arch Ophthalmol. 1995;113: 791–802.[pubmed]
[3].Albert DM. Historic review of retinoblastoma. Ophthalmology. 1987; 94:654–62. [pubmed]
[4].Schoeler D, Lindner T, Schulenburg S, Von der, Pink D, Reichardt P. Coincidence of retinoblastoma and leiomyosarcoma in father and daughter - a rare case report. ASCO Annual Meeting Proceedings (Post-Meeting Edition). J Clin Oncol 2004; 22 (14) (July 15 Supplement):9059.
[5].Shields JA, Shields CL. Retinoblastom: Clinical and pathologic features. In: Shields JA, Shield CL editors. Intraocular Tumors. A Text and Atlas, Philadelphia: WB Saunders Co; 1992;305-32.
[6].Magramm I, Abramson DH, Ellsworth RM. Optic nerve involvement in retinoblastoma. Ophthalmology. 1989; 96:217–22.[pubmed]
[7].Ellsworth RM. The practical management of retinoblastoma. Trans Am Ophthalmol Soc. 1969;67:462–534.[pubmed]
[8].White L. Chemotherapy in retinoblastoma current status and future directions. Am J Pediatr Hematol Oncol. 1991;13:189-201[pubmed]
[9].Shields CL, Honavar SG, Meadows AT, Shields JA, Demirci H, Singh A, Friedman DL, Naduvilath TJ. Chemoreduction plus focal therapy for retinoblastoma: factors predictive of need for treatment with external beam radiotherapy or enucleation. Am J Ophthalmol. 2002;133:657-64[pubmed]


