Research

Ligational Aspects of the Mesogenic Schiff-base, N,N’-di-(4-hexadecyloxy) salicylidenediaminoethane with some Transition Metal Ions
*Sanyucta Kumari
- Submitted: Friday, February 28, 2014
- Accepted: ; Sunday, March 30, 2014
- Published: Sunday, March 30, 2014
This is an Open Access article distributed under the terms of the Creative Commons Attribution License ((http://creativecommons.org/licenses/by/3.0)which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited
Abstract
A mesogenic Schiff-base, N,N’-di-(4-hexadecyloxy)salicylidenediaminoethane (H2L1) with smectic-C(SmC) and nematic(N) mesophases, was synthesized and its structure studied by elemental analyses and mass, NMR & IR spectra. The bi-dentate bonding of the Schiff-base in the mesogenic complex (LaIII), as implied on the basis of IR & NMR spectral data. As per the spectral studies of the complex, the Zwiterionic-species of the ligand, coordinates to the LnIII ion through two phenolateoxygens, rendering the overall geometry around Transition metl ion to distorted Square Anti-prism / Mono-capped Octahedron.
Keywords
Mesogenic Schiff-base, Square Anti-prism and Mono-capped Octahedron, crystal structure, NMR & IR spectra
Introduction
Liquid crystals with transition metal core groups are increasingly a topic of investigation since metals can impart useful shapes and properties which are not easily produced in totally organic liquid crystals [1]. A major distinction between metallomesogens and most organic mesogens is the greater tendency in the former type to exhibit intermolecular dative coordination in the solid state [1]. However, it should be noted that rigid tetrahedral molecules usually seem to prevent mesophaseformation[2]. As a part of our investigation on systematic structural and spectroscopic studies of 3d metal complexes of a series of mesogenic organic Schiff-bases, we report here, synthesis and spectroscopic studies on the mesogenic Schiff-base, N,N’-di-(4’-pentyloxybenzoate)salicylidene-l,8-diamino-3,6-dioxaoctane (H2L) and crystal structure of the corresponding Zn(II) complex. We have observed that despite the mesogenicbehaviour of the [CuL] (unpublished work), the zinc homologue, [ZnL], does not exhibit liquid crystalline properties presumably due to the tetrahedral coordination of the latter [2].
2. Experimental Section
2.1. Materials
All reagents were purchased fromcommercial sources and used as received: 1-bromohexadecane, 2, 4-dihydroxybenzaldehyde and 1, 2 diaminoethanearefrom Sigma-Aldrich, USA; all Ln(NO)3.xH2O, KIand KHCO3are from Merck. The solvents received were dried using standard methods [3] when required.
2.2. Synthesis and analysis
The synthesis of N,N’-di-(4-hexadecyloxy) salicylidenediaminoethane (H2L1) 2, was achieved by proceeding through two major steps, viz., alkylation of 2,4-dihydroxybenzaldehydewith n-hexadecyl bromide, followed by Schiff-base formation. All the experimental details are given in (scheme 1).[Mx(L1H2)3(NO3)y](NO3)2: (M =Sc,Ti,Vd,Cr,Zn; x = 1 or 2; y = 1 or 4) complexes were prepared by refluxing together hot dry ethanolic solutions of the ligand, H2L1, (2.25g, 3.0 mmol in 30 mL) and the appropriate metal nitrate (1.0 mmol, in 20 mL) for ~ 1h at 750C. The reaction mixture was left over night in the flask closed with guard tube.The solid complex that separated out in each case was filtered under suction and washed repeatedly with cold ethanol and dried over fused CaCl2 in a desiccator.
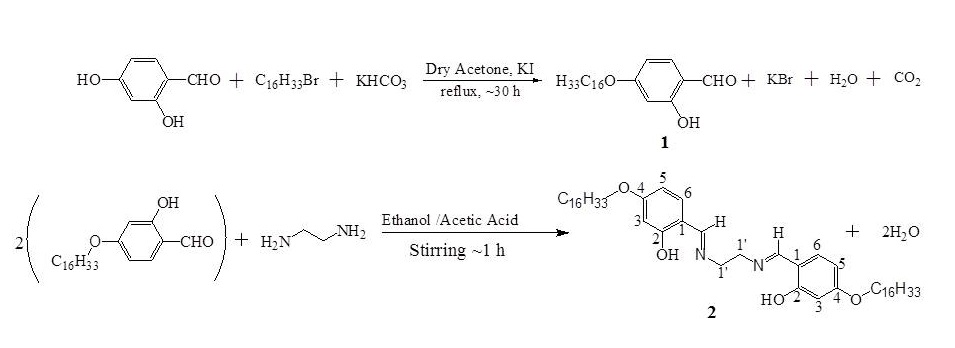
Scheme-1: Reaction steps involved in the synthesis of4-hexadecyloxysalicylaldehyde, 1and N, N’-di-(4-hexadecyloxy)salicylidenediaminoethane, H2dhdsde, 2, (H2L1).
2.2.1 Preparation of 4-octadecyloxybenzoic acid (1):
Equimolar amounts of 1-bromohexadecane (60mmol, 18.35 mL), 2,4-dihydroxy benzaldehyde (60mmol, 8.30 g) and potassium bicarbonate (66mmol, 6.68 g) were suspended in 250 ml of dried acetone and refluxed for ~30 h in presence of KI (0.1-0.2 g) as catalyst and filtered while hot to remove the insoluble solids, neutralized with dil. 6NHCl and extracted twice with CHCl3.The chloroform extracts were concentrated to obtain a purple solid which was then purified by column chromatography over SiO2 (by eluting with a mixture of n-hexane and chloroform (v/v, 1/1)); evaporation of this purified extract finally yielded 4-hexadecyloxysalicylaldehyde in the form of a white solid; yield: 80 % (17.39 g).
2.2.2 Synthesis of N, N’-di-(4-hexadecyloxy)salicylidenediaminoethane, 2:
The ligand, N,N’-di-(4-hexadecyloxy)salicylidenediaminoethane was prepared by refluxing together absolute ethanolic solutions of 4-hexadecyloxysalicyaldehyde (40 mmol, 14.48 g in 100 mL) and 1, 2 diaminoethane (20 mmol, 1.34 mL in 50 mL) for ~1h in presence of few drops of acetic acid and left over night in the flask closed with guard tube.The micro-crystalline yellow coloured product, 2, was suction-filtered, thoroughly washed with cold ethanol and recrystallized from a mixture of absolute ethanol and chloroforms (1:1 v/v) and dried at room temperature. Yield: 5.61 g (75 %); m.p. 100 0C.; 1H NMR(300 MHz; CDCl3; J(Hz), Me4Si at 25 °C, ppm) d = 0.875 (t, 3JH-H = 6.0 Hz, 3H, CH3), 1.778-1.232(m, 28H, Hmethylene),3.834(s, 2H, -NCH2),3.934(t, 3JH-H = 6.6 Hz, 2H, -OCH2), 6.342(s, 1H, H3), 6.381(d, 3JH-H = 6.0 Hz,1H, H5), 7.060(dd, 4JH-H = 1.2 Hz, 3JH-H = 8.4 Hz, 1H, H6,8.183(s, 1H, -N=CH), 13.655(s, 1H, Ph-OH); 13C{1H} NMR (75.45 MHz; CDCl3; Me4Si at 25 °C, ppm ) δ = 165.418(-C2), 164.635(-NCH), 163.127(-C4), 132.657(-C6), 112.169(-C1), 106.919(-C5), 101.628(-C3), 68.117(-OCH2) and 58.862(-NCH2);FAB Mass: The molecular ion (m/e, 749; 100% intensity) generates simultaneously two fragments, M1 – M2, (m/e, fragment, % intensity): M1: 164, HOC6H3(OH)CH=NCH2CH2+, 55;M2: 150, HOC6H3(OH)CH=NCH2+, 50%; IR (KBr, cm-1):n(O-H)phenol 3460br, n(C=N) 1628, n(C-O)phenol 1145; C48H80N2O4(749.16): Calcd. C 76.95, H 10.76, N 3.74; found C 76.92, H 10.71, N 3.71.
2.2.3 Synthesis of [Zn(L1H2)3(NO3)](NO3)2:
Anhydrous solutions of N,N’-di-(4-hexadecyloxy)salicylidenediaminoethane, 2,(2.25g, 3 mmol in 30 mL) in dichloromethane (30 mL) and of La(NO3)3.6H2O(0.433, 1 mmol in 20 mL) in ethanol were refluxed for ~ 6 h and the reaction mixture left over-night in the reaction falsk after reducing the volume to ~10 mL. The crude solid complex, was filtered off under suction, washed repeatedly with cold ethanol, recrystallised from the solution of chloroform/ethanol and dried over fused CaCl2 in a desiccator. Yield: 1.60 g (63 %); m.p. 200 0C.(decompose);1H NMR(300 MHz; CDCl3; J(Hz), Me4Si at 25 °C, ppm) d = 0.874 (t, 3JH-H = 8.1 Hz, 3H, CH3), 1.620-1.248(m, 28H, Hmethylene),3.450(s, 2H, -NCH2),3.772(t, 3JH-H = 6.8 Hz, 2H, -OCH2), 6.955(s, 1H, H3), 6.283(d, 3JH-H = 7.2 Hz,1H, H5), 7.251(d, 3JH-H = 8.7 Hz, 1H, H6),8.031(s, 1H, -N=CH), 12.317(br s, 1H, -N+H);13C{1H} NMR (75.45 MHz; CDCl3; Me4Si at 25 °C, ppm ) δ = 165.327(-C2), 178.514(-NCH), 1583602(-C4), 135.921(-C6), 133.679(-C1), 108.888(-C5), 108.625(-C3) and 68.101(-OCH2); IR (KBr, cm-1):n(C=N) 1648, n(C-O)phenol 1122; LaC144H240N9O21 (2572.40): Calcd. C 67.23, H 9.40, N 4.90, La 5.40; found C 67.27, H 9.35, N 4.91, La 5.38.
2.2.4 Synthesis of [Ti2(L1H2)3(NO3)4](NO3)2:
Yield: 1.79 g (61 %); m.p. 200 0C.(decompose);IR (KBr, cm-1): IR (KBr, cm-1):n(C=N) 1647, n(C-O)phenol1128; Gd2C144H240N12O30(2934.01): Calcd. C 58.95, H 8.24, N 5.73, Gd 10.72; found C 59.01, H 8.23, N 5.75, Gd 10.69.
2.2.5 Synthesis of Vd2(L1H2)3(NO3)4](NO3)2:
Yield: 1.71 g (58 %); m.p. 200 0C.(decompose); IR (KBr, cm-1): n(C=N) 1647, n(C-O)phenol 1128; Ho2C144H240N12O30(2949.37): Calcd. C 58.64, H 8.20, N 5.70, Ho 11.18; found C 58.60, H 8.17, N 5.66, Zn 11.16.
2.3 Physical measurements
The 1H and 13C NMR spectra were recorded on a JEOL AL-300 MHz FT-NMR multinuclear spectrometer; C, H, and N contents were micro analyzed on Elemental Vario EL III Carlo Erba 1108 analyzer. Infrared spectra were recorded on JASCO FT/IR (model-5300) spectrophotometer in the 4000-400 cm-1 region. The mass spectra were recorded on JEOL SX-102 FAB mass spectrometers. The UV-VIS spectra were recorded on Shimadzu spectrophotometer, model Pharmaspec- UV 1700. Magnetic susceptibility measurements were made at room temperature on a Cahn-Faraday balance.
2.4. X-ray Crystallographic Data Collection and Refinement of the Structure
X-ray data for the compound H2L1, [aa53m],was collected at room temperature using a Bruker Smart Apex CCD diffractometer with graphite monochromatedMoKa radiation (l=0.71073Å) with ω-scan method. Preliminary lattice parameters and orientation matrices were obtained from four sets of frames. Unit cell dimensions were determined from the setting angles of 2421 reflections in the range of 2.59
Integration and scaling of intensity data was accomplished using SAINT program [1]. The structure was solved by direct methods using SHELXS97 [2] and refinement was carried out by full-matrix least-squares technique using SHELXL97 [2]. Anisotropic displacement parameters were included for all non-hydrogen atoms. The hydrogen atom attached to the oxygen atom was located in difference Fourier map and their positions and isotropic displacement parameter were refined. All other hydrogen atoms were positioned geometrically and treated as riding atoms, with C-H distances in the range of 0.93-0.96Å and with Uiso(H) values of 1.5Ueq( C ) for methyl hydrogen and 1.2Ueq(C) for other hydrogen atoms.
3. Results and Discussion
3.1. Spectral investigation
The Schiff-base ligand (H2L1), 2, is light yellow-coloured while other LnIIIcomplexes, are cream or white coloured. Both the parent ligand and the metal complexes are soluble in chloroform, dichloromethane, DMF and DMSO but are insoluble in water, methanol, ethanol, and acetonitrile. The structures and purities of the Schiff-base ligand, 2, and of the metal complexes, were confirmed by IR and 1H NMR spectroscopy and elemental analyses. The structure of the ligand was further confirmed by FAB Mass spectrum. The mass spectral features of H2L1were characterized by the base peak as well as molecular ion peak corresponding to the m/e value of 749, which matches with the molecular weight of 749.61 of H2L1 of the molecular formula, C48H80N2O4.The 100% intensity of the molecular ion peak is as expected for the molecule on the basis of its predominant aromatic character; further, the 100% intensity of the molecular ion peak (i.e., the base peak) of m/e = 749, is usually considered as an excellent diagnostic peak and the major fragment peaks (m/e = 164, 150) due to HOC6H3(OH)CH=NCH2+ and HOC6H3(OH)CH=NCH2CH2+.A comparison of the 1H and 13C{1H}NMR spectral data of the ligand with that of the LaIII complex shows the absence of the phenolic-OH signal. From the composition of the LaIIIcomplex,[La(L1H2)3(NO3)](NO3)2, it willappear that the ligand (H2L1) is acting as a neutral ligand;however, the 1H NMR spectrum of the compound has revealed that the phenolic protons are shifted to the twouncoordinated iminonitrogens, which then get intramolecularlyhydrogen-bonded to the metal-bound phenolateoxygens to giverise to the zwitterionic structure = +N–H• • •O–.The macrocycle under this condition is designated as L1H243. We found that the signal correspondingto the imine hydrogen, -CH-N, was broadened in theLaIII complex (d, 8.031) when compared with the samesignal in the Schiff base ligand (d, 8.183) complex of LaIII, Further,a new signal appears at d, 12.317, characteristic of –N+H resonance appears in the spectrum of the LaIII complex of H2L1 while the parent ligand does not show any such signal. These observations imply that the Schiff base is present in a zwitterionicform,with the phenolic oxygen deprotonated and the iminenitrogen protonated (scheme. 2).
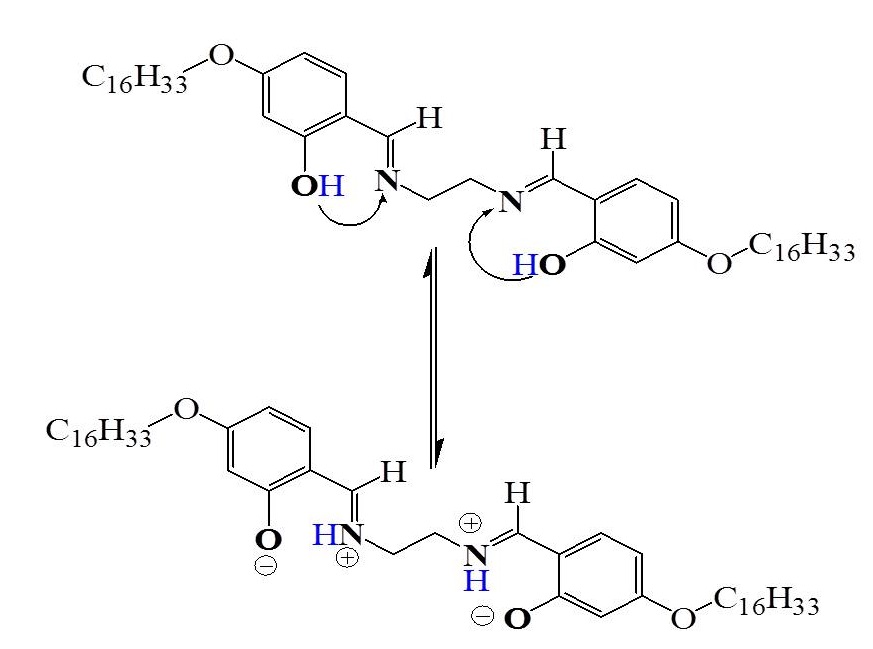
Scheme 2: Depiction of migration of phenolic protons to imine nitrogens of the ligand, H2L1, during the formation of zwitter ion
Binnemanset al51, while reporting their work on rare earth-containing magnetic liquid crystals of the formula, [Ln(LH)3(NO3)3], where LH = 4-alkoxy-N-alkyl-2-hydroxybenzaldimine,found that selective irradiationof the signal at d, 12.29 ppm removed the broadening of the imine signal,thereby inferring that the signal does not correspond to the protonof the -OH group, but to the proton of the -N+H group. Further, by variable temperature 1H NMR spectra, they showed that at 293 K, the value of the coupling constant3J = 10.3 Hz pointed to a trans-disposition of the –CH= and =N+H- protons andan increase in the temperature lead to abroadening of the signal of the -CH=N proton in the ligand from4.1 Hz at 293 K to 13.7 Hz at 333 K. This provides sufficient experimental evidence for showing the formation of the zwitter ion. The single crystal structure solved by the same authors51 was also in agreement with the zwitter ionic form of the ligand within the complexes.
The 13C{1H} NMR spectra show a significant shift of the -NCH signal from d, 164.635 (in the case of H2L1) to d, 178.514 (in the case of the LaIII complex). Similar shifts observed in the case of the carbon atoms directly attached to the bonding atoms (phenolate carbons) while those for the other carbons were of lesser magnitude. Thus, the NMR spectral data imply bonding through the two phenolateoxygens of the ligand in the zwitter ionic form to the LaIII metal ion.
The IR spectra (recorded as KBr pellets) of the parent ligand (H2L1) and of the metal complexes; The broad absorption centered on 3460 cm-1 may be assigned to the O-H stretching vibration of the phenolic group37, may be understood to involve considerable amount of H-bonding (presumably intramolecularly bonded to the ortho>C=N group) under the experimental conditions. The n(O-H)phenolic band totally disappears in the spectra of the complexes due to the shifting of the phenolic proton to the azomethine nitrogen atom resulting in the formation of zwitter ion. The weak/medium intensity bands centered on 1180 cm-1 due to C-O stretching modes of phenolic group. The strong intensity band occurring at 1628 cm-1 may be assigned44 to n(C=N) absorption of the azomethine moiety.
This band undergoes a hypsochromic shift in all the complexes on account of the formation. The process of complexation of the ligand with LnIII ions, resulted in the shifting of the phenolic protons to the two uncoordinated iminonitrogens, which then get intramolecularly hydrogen-bonded to the metal-bound phenolateoxygens to give rise to the zwitterionic structure, =+N–H• • •O−. Similar zwitterionicbehaviour has been reported by others for acyclic Schiff base lanthanide complexes47. The formation of a zwitterionicform can be rationalized by the tendency of the lanthanide ions to coordinate to negatively charged ligands with a preference for O-donor ligands. By transfer of the phenolic proton to the imine nitrogen, the phenolic oxygen becomes negatively charged and can coordinate to the lanthanide ion. The initial evidence for zwitter ionic formation was obtained from 1H NMR spectra. Further evidence for the existence of a zwitter-ionic form in the metal complexes was given by infrared spectroscopy and, more particularly, by the band frequencies of the C=N vibration. The shift to higher wavenumbers in the complexes compared to the corresponding values in the ligands indicates that the nitrogen atom is not involved in the complex formation and that a C-N+ group is present48.
Further, all the complexes are characterized by a strong band due to n(C=N) at 1645(8) cm−1 and a weak broad band at about 3040(20) cm−1 due to the hydrogen bonded N–H • • •O vibration of the protonated imine43. The ligands coordinate to the metal ion via the negatively charged phenolic oxygen only. No binding occurs between the lanthanide ion and the imine nitrogen, and the three nitrate groups coordinate in a bidentate fashion.The LnIII complexes also exhibit three additional bands around 1469–1466, 1285–1282, and 795-790 cm-1, which can be assigned to the vibrational modes of the coordinated (C2v) nitrate groups49. The magnitude of splitting at higher energies, 187–181 cm-1, suggests that the coordinated nitrate groups act as bidentate ligands49,50. The additional bands observed at 1383–1374 cm-1 are due to the non-coordinated nitrate present in the complexes49,50. At this juncture, it may be recalled that complexes of two different M:L ratios are formed within this series, viz., [Ln(L1H2)3(NO3)](NO3)2 and [Ln’2(L1H2)3(NO3)4](NO3)2 where Ln = La, Pr, Nd, Sm and Eu and Ln’ = Gd, Tb, Dy and Ho; ionic as well as bidentate-coordinate nitrate groups are present in both the types of complexes. Distorted square-antiprism with C.N. = 8 and mono-capped octahedron with C.N. = 7 have been proposed respectively for the first half and second half of the series of the complexes.
The asymmetric unit contains one half-molecule with the other half generated by the crystallographic inversion center that passes through the ethylene bridge. Geometrical isomerism around the double bond C23=N1 affords the possibility of cis and trans isomers. The present structure is the E isomer, as shown by the C1-C6-C23-N1 torsion angle of -1.5(4)°. The salicyidene six-membered ring is planar. For the hexadecyl chain C7-C22, the average C-C bond length and C-C-C angles are 1.515(3) Å and 114.0(3)° respectively. The hexadecyl chain with torsion angles ranging from 178.0(2)° -179.8(2)° defines a near trans-coplanar conformation. Intramolecular O-H…N hydrogen bond is observed in the crystal structure and forms pseudo six-membered ring of a characteristic S(6)-type motif [3].
The Schiff base ligand is a liquid crystal itself, displaying a smectic C (SmC), and a nematic (N) phase (Crys • 75.63 •SmC • 93.72 • N • 101.93 • I) (see Table1). The mesophases were identified by the defect textures observed by polarized hot-stage microscopy. Typical for the nematic phase is the Schlieren texture with two and four brushes. The nematic phase separates from the isotropic liquid as droplets. The smectic C phase could be identified unambiguously by the texture change and the SmC« N transition for which a transient worm-like texture was observed. The viscosity of the smectic C phase was quite low, and as in the case of the nematic phase, Brownian motion of the molecules was visible. The mesophasebehaviour is different in the sense that no smectic phase except LaIII complex (show smectic X phase) is observed for the lanthanide complexes (only a nematicphase).This can easily be observed by polarized hot-stagemicroscopy and DSC [Fig 1] . Moreover, the lanthanidecomplexes do not show a clearing point, but they decompose in the mesophase.
Table 1. Mesomorphism of the Schiff base ligand, H2L1and the corresponding lanthanide complexes
|
Compound
|
Transition Temperature (°C)
|
|
H2L1
|
Crys· 75.63 ·SmC· 93.72 · N· 101.93 · I
I · 100.75 · N· 92.41 ·SmC· 70.52 ·Crys
|
|
[Ti(L1H2)3NO3](NO3)2
|
Crys· 83.80 ·SmX·155.48·N· 198.58·dec.
|
|
[Vd(L1H2)3NO3](NO3)2
|
Crys· 203.97·N· 248.48·dec.
|
Abbreviations: Crys = Crystalline phase, SmC = Smectic C phase, SmX = Smectic X phase, N = Nematic phase, I = Isotropic liquid, dec. = decomposition
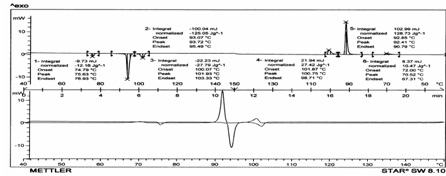
Figure 1. DSC thermogram of Schiff base ligand, H2L1; endothermic peaks are pointing downwards and exothermic peaks are pointing upwards.
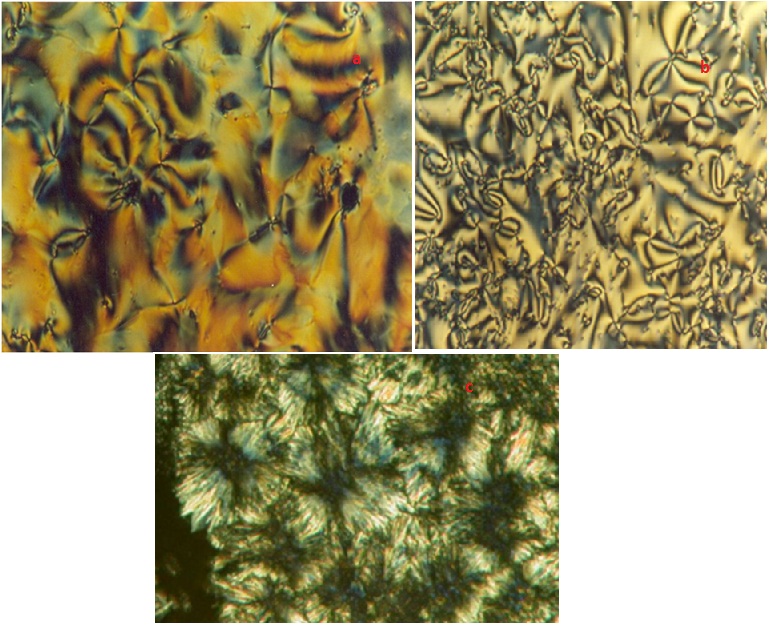
Figure 2(a) H2L1, Fig. (b) [Ti(L1H2)3NO3](NO3)2, Fig. (c) [Vd(L1H2)3NO3](NO3)2
4. Conclusions
The Schiff-base, N,N’-di-(4’-pentyloxybenzoate)salicylidene-l,8-diamino-3,6-dioxaoctane (H2L4) with the mesogenic phase, nematic droplets, coordinates to Zn(II) as a di-negative tetra-dentate species, to yield the non-mesogenic tetrahedral complex, [ZnL4]. As per the crystal structure of the complex, the dinegative species of the ligand, L42-, coordinates to the Zn(II) ion through two phenolateoxygens and two azomethinenitrogens, rendering the overall geometry around Zn(II) to distorted tetrahedron.
Acknowledgements
The authors wish to acknowledge the recording of FAB Mass spectra and elemental analyses at the Central Drug Research Institute, Lucknow. One of the authors, T. R. Rao, gratefully acknowledges the financial grant received from the Council of Scientific & Industrial Research, New Delhi [vide grant No. 01(1834)/03/EMR-II dated 12-03-2003] and the University Grants Commission [vide grant No. F.12-26/2003 (SR) dated 31 Mar 2003].
Conflict of interest
None
Ethical Consideration
None
Funding
None
References
[1].Serrette, P. J. Carroll, T. M. Swager, J. Am. Chem Soc. 114(1992) 1887. (b) A. M. Giroud-Godquin, P. M. Maitlis, Angew. Chem. Int. Ed. Engl. 30 (1991) 375.
[2].Pegenau, T. Hegmann, C. Tschierske, S. Diele, Chem. Eur. J. 5 (1999) 1643.
[3].K. Binnemans and K. Lodewyckx, Angew. Chem,. 2001, 113,248-250.[pubmed]
[4].Piechocki, J. Simon, J.J. Andre, D. Guillon, P. Petit, A. Skoulios, P. Weber, Chem. Phys. Lett. 122 1985 124.
[5].Yu.G. Galyametdinov, G.I. Ivanova, I.V. Ovchinnikov, Bull. Acad., Sci. USSR, Div. Chem. Sci. 40 1991 1109.
[6].A.Weisberger and F.S.Praskver ‘Organic solvents’ International publishers Inc., New York(1956) 1263.
[7].SMART & SAINT. Software Reference Manuals. Versions 6.28a & 5.625, BrukerAnalytical X-ray Systems Inc., Madison, Wisconsin, U.S.A., 2001.
[8].Sheldrick, G. M. SHELXS97 and SHELXL97, Programs for crystal structure solution and refinement; University of Gottingen: Germany, 1997.
[9]K. Binnemans, K. Lodewyckx, R. van Deun, Y.G. Galyametdinov, D. Hinz, and G. Meyer, Liquid Crystals. 28 (2001) 279.
[10].R. M. Silverstein, F. X. Webster, ‘Spectroscopic Identification of Organic Compounds’, John-Wiley & Sons. Inc., New York, 6th Ed., (2002) 82-98.
[11]. A. Braibanti, F. Dallavalle, and M. A. Pellinghelli, Inorg. Chem.7 (1968) 1430.

